Trillium Diagnostik 2018; 16(4): 278-281
Autoinflammatorische Erkrankungen, wie z. B. das familiäre Mittelmeerfieber oder das Cryopyrin-assoziierte periodische Syndrom, können durch eine gestörte Aktivierung des angeborenen Immunsystems zu einer überschießenden Entzündungsreaktion führen. Mutationen im Erbgut führen zu veränderten Genprodukten, und diese wiederum zur Produktion pro-inflammatorischer Zytokine. Durch Fortschritte in der Genetik konnten neue hocheffektive Therapien für bisher kaum behandelbare Erkrankungen gefunden werden.
Schlüsselwörter: Inflammasom, Pyrin, Cryopyrin, CAPS, TRAPS, MKD, PAPA-Syndrom
Unter dem Begriff Autoinflammatorische Syndrome (autoinflammatory diseases, AID) werden eine Reihe von erblichen und nicht-erblichen Erkrankungen zusammengefasst, die sich pathophysiologisch v. a. durch eine gestörte Aktivierung des Immunsystems und einer damit verbundenen überschießenden Entzündungsreaktion auszeichnen. Sie manifestieren sich meist bereits im Kindesalter. Ursache ist z. B. eine gesteigerte Ausschüttung von pro-inflammatorischen Zytokinen wie IL-1. Klinisch treten häufig rezidivierende Fieberschübe und ausgeprägte serologische Entzündungszeichen, die u. a. zu Symptomen an Haut, serösen Häuten, Schleimhäuten und Gelenken führen, auf. Daher spielen diese Erkrankungen in der Differenzialdiagnose des Fiebers unklarer Genese eine wichtige Rolle – insbesondere, wenn es sich um rezidivierende Fieberschübe handelt.
Das („unspezifische“) Immunsystem bietet einen sofortigen Schutz gegen sogenannte Gefahrensignale („danger signals“). Eine Vielzahl von Rezeptoren erkennt die Erreger anhand von hoch konservierten Strukturen ihrer Zellmembran, welche man als PAMPs („pathogen-associated molecular pattern“) bezeichnet. Auch endogene Gefahrensignale (sog. Alarmine), z. B. fehlerhaft gefaltete Proteine, sind erkennbar. Unter dem übergeordneten Begriff „damage-associated molecular patterns“ (DAMPs) fasst man alle diese Trigger zusammen, die durch die Bindung an spezielle Rezeptoren beispielsweise zur Zytokinsekretion oder zum Zelluntergang (Apoptose) führen.
Die Rezeptoren sind als „Toll-like Rezeptoren“ (TLR) in der Zellmembran oder wie z. B. die sog. NLR („nucleotide-binding domain, leucine-rich family“) auch intrazellulär lokalisiert. Zu diesen gehören eine Reihe von Proteinen wie NOD („nucleotide binding and oligorimerisation domain“) und NTPasen (Nukleosid-Triphosphatasen).
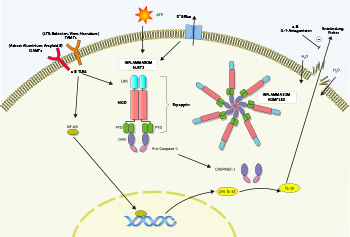
Zytoplasmatische Komplexe, die meist aus vielen dieser Proteine aufgebaut sind und eine wichtige Rolle in der Entzündungsreaktion spielen, werden als „Inflammasom“ bezeichnet (Abb. 1). Ein typisches Inflammasom besteht aus einem Rezeptor, mehreren Adapterproteinen und einem oder mehreren inflammatorischen Proteasen (Caspasen). Das Inflammasom kann durch exogene und endogene Schadstoffe aktiviert werden, beispielsweise durch Endotoxin bei bakteriellen Infektionen oder Harnsäurekristalle bei Gichtpatienten.
Viele AID werden durch Mutationen in Genen von Proteinen, die am Aufbau von Inflammasomen beteiligt sind, verursacht. Die mutierten Proteine führen dabei oft zu einer Autoaktivierung des Inflammasoms und zu einer Überproduktion von IL (Interleukin)-1β (Tab. 1) [1, 2].