Die Typ-1-Interferonopathien stellen eine Gruppe genetisch bedingter seltener Erkrankungen dar, die durch eine Fehlfunktion des angeborenen Immunsystems hervorgerufen werden. Ihr klinisches Spektrum ist sehr breit und sowohl durch Autoinflammation als auch durch Autoimmunität charakterisiert. Gemeinsames Kennzeichen ist eine Dysregulation der antiviralen Typ-1-Interferon (IFN)-Achse, die zu einer konstitutiven Typ-1-IFN-Aktivierung führt. Pathogenetisch liegen den Typ-1-Interferonopathien Störungen im Metabolismus und im Sensing von intrazellulären Nukleinsäuren zugrunde. Unser derzeitiges Wissen über die Entstehung der Typ-1-Interferonopathien weist darauf hin, dass eine Hemmung der pathogenen Typ-1-IFN-Aktvierung therapeutisch wirksam sein könnte.
Schlüsselwörter: Typ-1-Interferon, Autoinflammation, Autoimmunität, angeborene Immunität, Genetik, Pathogenese
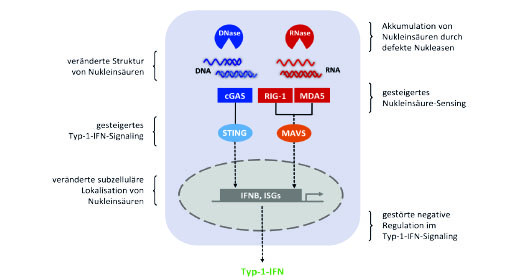
Typ-1-Interferone, IFN-α und IFN-β, fungieren als wesentliche Effektorzytokine der Immunantwort auf Viren und andere intrazelluläre Erreger. Die Produktion von Typ-1-IFN, zu der fast alle Körperzellen befähigt sind, erfolgt normalerweise nicht konstitutiv, sondern wird durch Rezeptoren des angeborenen Immunsystems induziert, welche Gefahrensignale ausgehend von pathogenen Erregern detektieren. Die Aktivierung der Typ-1-IFN-Signalkaskade resultiert in der transkriptionellen Induktion hunderter IFN-stimulierter Gene, deren Zusammenspiel den Wirtsorganismus in einen antiviralen Zustand versetzt, mit dem Ziel, infizierte Zellen zu eliminieren und die Verbreitung einer Infektion zu limitieren (Abb. 1). Eine unkontrollierte Aktivierung von Typ-1-IFN kann jedoch schädliche Folgen für den Wirtsorganismus haben, da dies inadäquate Entzündungsprozesse sowie den Verlust der immunologischen Toleranz begünstigt. Die Aufklärung der genetischen Ursachen seltener monogener Krankheitsbilder, die mit einer chronischen Typ-1-IFN-Aktivierung einhergehen, hat zur Identifizierung neuer Krankheitsmechanismen beigetragen, die zu Autoinflammation und Autoimmunität führen.
Nukleinsäure-Sensing und Typ-1-Interferon
Die Wahrnehmung einer viralen Infektion durch den Wirtsorganismus erfolgt primär über die Detektion viraler Nukleinsäuren, die als fremde mikrobielle Strukturen (Pathogen-associated molecular pattern) von Mustererkennungsrezeptoren (Pattern recognition receptor) erkannt werden. Im Endosom dendritischer Zellen werden RNA und DNA, welche aus phagozytierten viralen Partikeln stammen, durch Toll-like Rezeptoren TLR3, TLR7 und TLR9 registriert. Die TLR-Aktivierung führt vermittelt über die Transkriptionsfaktoren IRF3, IRF7 und NK-κB zur Induktion von Typ-1-IFN und pro-inflammatorischer Zytokine [1]. Im Zytosol hingegen erfolgt die Erkennung von Nukleinsäuren TLR-unabhängig (Abb. 1). So wird zytosolische RNA durch zwei ubiquitär exprimierte Rezeptoren, RIG-I und MDA5, erkannt. Während RIG-I kurze dsRNA mit einem Triphosphat am 5´-Ende bindet, stellen lange dsRNA-Moleküle die Liganden von MDA5 dar. Beide RNA-Sensoren aktivieren NK-κB und IRF3/IRF7 nach Rekrutierung an das MAVS-Adapterprotein [2]. Das Sensing von zytosolischer DNA erfolgt im Wesentlichen durch die Nukleotidyltransferase cGAS, die nach Bindung von DNA die Synthese des Second Messengers cGAMP katalysiert. Die Rekrutierung von cGAMP an das Adaptermolekül STING aktiviert IRF3, welches in den Kern transloziert und die Transkription des IFNB-Gens induziert [2].
Die biologischen Funktionen von Typ-1-IFN werden über den Typ-1-IFN-Rezeptor vermittelt, einem Oberflächenrezeptor bestehend aus den zwei Untereinheiten IFNAR1 und IFNAR2 [3]. Die Bindung von Typ-1-IFN an seinen Rezeptor setzt eine Signalkaskade in Gang, die mit der Phosphorylierung der Rezeptor-assoziierten Janus-Kinasen JAK1 und Tyrosinkinase 2 initiiert wird. Dies ermöglicht die Bindung der Transkriptionsfaktoren Signal transducers and activators of transcription 1 (STAT1) und STAT2 an den Rezeptor. Die nachfolgende Phosphorylierung von STAT1 und STAT2 induziert deren Dimerisierung und Translokation in den Zellkern, wo sie die Expression von IFNB sowie zahlreicher IFN-stimulierter Gene aktvieren [3]. Auf diese Weise beeinflussen Typ-1-IFN im Rahmen der antiviralen Immunantwort verschiedenste zelluläre Prozesse, die einerseits pro-apoptotische, andererseits auch immunostimulatorische und pro-inflammatorische Effekte ausüben.