Schlüsselwörter: mtDNA, nDNA, Stufendiagnostik, Next Generation Sequencing, RNA-Sequenzierung
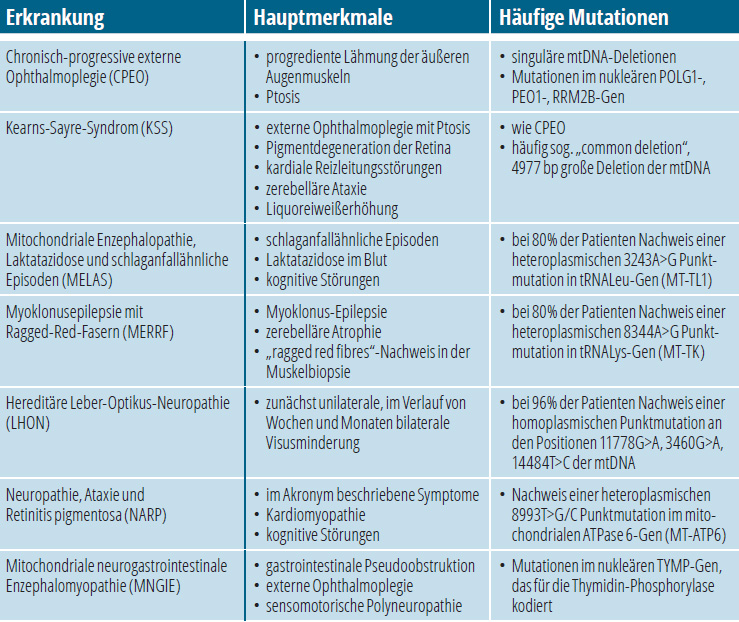
Mitochondrien spielen eine wichtige Rolle bei verschiedenen zellulären Prozessen des Stoffwechsels. Funktionsstörungen der Mitochondrien betreffen insbesondere Zellen, die einen hohen Energiebedarf haben, z. B. Muskel- und Nervenzellen, und können durch Defekte des Energie-generierenden Systems oder auch in zahlreichen Signal- und Stoffwechselwegen ausgelöst werden. Die sogenannten Mitochondriopathien sind vielgestaltige Erkrankungen, die eine große phänotypische, biochemische und genetische Heterogenität aufweisen und vielfältige genetische Ursachen haben. Sie können je nach Erkrankung sowohl im Kindes- und Jugendalter als auch – beispielsweise infolge einer Akkumulation somatischer Mutationen – beim Erwachsenen auftreten. Typische Zeichen, die einer mitochondrialen Dysfunktion zugeordnet werden können, lassen sich sowohl bei systemischen Erkrankungen als auch bei Krankheiten der unterschiedlichsten Organe (Muskel, Gehirn, Nerven, Niere, Herz, Leber, Augen, Ohren, Pankreas) finden [1]. In Tabelle 1 ist eine Auswahl von mitochondrialen Erkrankungen, deren typischer Phänotyp und häufig beobachtete Mutationen aufgeführt.
Mit einer Prävalenz von ca. 1 : 5.000 sind Mitochondriopathien gar nicht so selten wie bisher angenommen. Dennoch ist die Diagnosestellung aufgrund einer fehlenden Korrelation zwischen Genotyp und Phänotyp sehr aufwendig und stellt bis auf wenige charakteristische Syndrome immer noch eine große Herausforderung dar. Hinzu kommt, dass auch nach erfolgreicher Diagnostik die weitere Behandlung der Patienten oft nur symptomatisch möglich ist, da bei vielen dieser Erkrankungen kurative Therapieoptionen fehlen.
Genetische Grundlagen
Bereits 1890 beschrieb der Anatom Richard Altmann die Mitochondrien als subzelluläre Bestandteile, sog. „cytoplasmatische Organellen“, welche er als „Elementarorganismen der Zelle“ bezeichnete [2]. Erst viel später – zu Beginn der 1980er-Jahre – wurde die weitgehend maternale Vererbung der mitochondrialen DNA (mtDNA) nachgewiesen und das menschliche mitochondriale Genom entschlüsselt [3, 4]. Die zirkuläre doppelsträngige mtDNA im Inneren der Mitochondrien mit einer Größe von 16.569 Basenpaaren weist eine weitaus höhere Mutationsrate auf als die nukleäre DNA (nDNA), da Mitochondrien über weniger Reparatursysteme verfügen und die mtDNA nicht durch Histone geschützt wird. Mit der Entdeckung genetischer Veränderungen im mitochondrialen Genom gegen Ende der 1980er-Jahre und deren Assoziation mit Erkrankungen wurde ein neues medizinisches Spezialgebiet erschlossen, dessen Vielfältigkeit seither ständig gewachsen ist.
Kennzeichnend für krankheitsverursachende Mutationen der mtDNA ist deren Heteroplasmie. Dabei können in einer Zelle – aber auch innerhalb eines Mitochondriums – gemischte Populationen von mtDNA-Kopien mit und ohne Veränderungen vorliegen. Überschreitet der Anteil von mutierter mtDNA einen gewissen Schwellenwert, kommt es meistens zum Auftreten von Symptomen. Der Heteroplasmiegrad (prozentualer Anteil mutierter mtDNA-Kopien) schwankt in unterschiedlichen Geweben und bestimmt maßgeblich Ausprägung und Verlauf der Erkrankung. Entsprechend geht ein hoher Heteroplasmiegrad oft mit einem schweren Krankheitsverlauf einher.
Noch häufiger sind Kerngenom-Mutationen Ursache von Mitochondriopathien. Dabei sind u. a. Komponenten der mitochondrialen Replikation, Transkription oder Translation, Kofaktoren, Assemblierungsfaktoren oder Strukturproteine der Mitochondrien betroffen. Im humanen Genom befinden sich nach heutigem Stand 1.158 Gene, die für mitochondrial-lokalisierte Proteine kodieren [4]. Da jedes dieser Gene ein potenzielles Kandidatengen darstellt, gestaltet sich die Diagnostik entsprechend anspruchsvoll. Im Gegensatz zu den Mitochondriopathien im Erwachsenenalter, die überwiegend durch Veränderungen in der mtDNA selbst bedingt sind, finden sich bei den kindlichen Erkrankungen häufiger Mutationen der nDNA. Derzeit sind mehr als 330 putative Krankheitsgene bekannt.
Patienten, die mtDNA-Mutationen aufweisen, erkranken häufig erst nach Jahren oder Jahrzehnten, was mit einer Akkumulation von mtDNA-Mutationen auch aufgrund fehlender Reparaturmechanismen zu erklären ist. Mit steigendem Alter kommen weitere zufällige Mutationen hinzu, die sich dann in den entsprechenden Geweben manifestieren und nach Überschreiten eines Schwellenwertes symptomatisch werden können. Mittlerweile mehren sich auch die Indizien für eine Beteiligung somatischer mtDNA-Mutationen und damit mitochondrialer Dysfunktionen bei zahlreichen altersbedingten Erkrankungen [6].