Die Diagnostik von erblichen Interferonopathien ist weltweit bisher wenigen Zentren vorbehalten. Das liegt vorrangig am Fehlen von Routinelaborparametern, die eine Erhöhung von Typ-I-Interferonen anzeigen, und an der Unkenntnis der seltenen Erkrankung mit klinisch variablen Beschwerdebildern. Diese Probleme bei der Diagnosestellung sind jedoch hochrelevant, da in der Rheumatologie zunehmend Typ-I-Inhibitoren verfügbar werden, die eine klinische Besserung in Aussicht stellen. Wir beschreiben hier den Fall eines 49-jährigen Patienten, der nach jahrzehntelangen klinischen Beschwerden die Diagnose einer erblichen Interferonopathie, des Singleton-Merten-Syndroms, erhalten hat, und kommentieren anschließend die Diagnostik einer erblichen Interferonopathie und deren Behandlungsmöglichkeiten.
Schlüsselwörter: Singleton-Merten-Syndrom, Interferonopathie, SIGLEC1
Krankheitsgeschichte
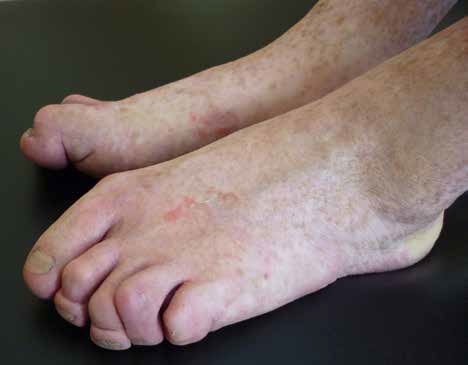
Im Folgenden wird der lehrreiche und zugleich problematische Fall eines nunmehr 49-jährigen Mannes vorgestellt, der nach über vier Jahrzehnten und zahllosen Vorstellungen in den Fachrichtungen Orthopädie, Dermatologie, Zahnmedizin, Rheumatologie und Humangenetik eine gesicherte Diagnose gestellt bekommen hat. Einen ersten klinischen Eindruck des Patienten vermittelt Abbildung 1, die klinische Hauptbefunde zeigt.
Im Alter von fünf Jahren führte die orthopädische Vorstellung zur Diagnose von Plattfüßen (Pes plano valgus). Bei einer späteren Wiedervorstellung im Alter von 13 Jahren wurden zusätzlich eine verstärkte Innenrotation der Hüfte, X-Beine (Genu valgum bds.) und eine Tibiatorsion beidseits beschrieben. Das damit verbundene veränderte und auffällige Gangbild mündete zudem in dem Verdacht auf eine infantile Zerebralparese, der sich jedoch nie bestätigte.
Bei Hautveränderungen an den Fußinnenflächen diagnostizierte ein Hautarzt im Alter von sechs Jahren eine Schuppenflechte (Psoriasis). Seit Kindheit bestehen zudem sehr viele und über die gesamte Haut verteilte braune, linsenförmige Flecken, die im Volksmund als Altersflecken bekannt sind. Diese durch Hautärzte als Lentigo bezeichneten Flecken sind bei fast allen beschriebenen Patienten (altersabhängig) dokumentiert.
Ab dem Alter von 11 Jahren kam es zu sehr häufigen zahnärztlichen Vorstellungen, da einerseits die regelrecht angelegten Milchzähne bis zum 30. Lebensjahr nicht ausfielen, und andererseits die dauerhaften zweiten Zähne nicht durchbrachen. Ein im jugendlichen Alter angefertigtes Übersichtsröntgen des Gebisses zeigte regelrecht angelegte zweite Zähne. Röntgenaufnahmen im Verlauf dokumentierten die Resorption der Zahnwurzeln der dauerhaften Zähne. Nach zahlreichen Zahnextraktionen der über die Zeit abgeschliffenen Milchzähne erfolgte im Alter von 30 Jahren die totale operative Gebiss-Extraktion und das Einsetzen einer Totalprothese. Die Anodontie, das vollständige Ausbleiben einer Zahnentwicklung, tritt am häufigsten im Rahmen von genetischen Syndromen auf.
Das Ausbleiben der zweiten Zahnung liefert in diesem Fall den spezifischsten Hinweis auf eine genetische Ursache der Beschwerden.
Im Alter von 34 Jahren stellt sich der Patient erstmalig in der Rheumatologie zur Abklärung einer fortschreitenden Hypermobilität der Hand- und Fußgelenke vor. Die am meisten ausgeprägten Veränderungen fanden sich an den Fingergrundgelenken (Metacarpophalangeal-Gelenke) beider Hände. Die Röntgenaufnahmen zeigten als einzige knöcherne Veränderungen die Auflösung der letzten Knochenglieder an Händen und Füßen (Akroosteolysen) sowie eine reduzierte Knochensubstanz (Osteopenie). Zeitgleich kam es innerhalb von ungefähr einem Jahr jeweils nach Stolpern zu Quadrizepssehnenrupturen beidseits, die operativ versorgt werden mussten. Die fortschreitenden Gelenkveränderungen wurden als Psoriasis-Arthritis gewertet und mit Methotrexat behandelt, welches jedoch aufgrund von Blutbildveränderungen (Leukopenie, Lymphopenie) wieder abgesetzt wurde. Im Verlauf wurde der Patient letztendlich auf Ustekinumab, einen humanen Antikörper, der die Zytokine Interleukin-12 und Interleukin-23 blockt und seit 2009 für die Psoriasis zugelassen ist, eingestellt. Darunter zeigten sich zwar die als Psoriasis klinisch klassifizierten Hautveränderungen regredient, die Gelenkveränderungen waren jedoch stetig progredient. Die radiologischen Veränderungen sind in den Abbildungen 2 und 3 zusammengetragen.
Bezüglich der Blutbildveränderungen berichtete der Patient, dass bereits Jahre vor der Methotrexat-Behandlung wiederholte Laboruntersuchungen eine Leuko- und Lymphopenie aufgezeigt hätten. Neben zahlreichen anderen Ursachen für einen Mangel an Blutzellen sind virale Infektionen oder systemische Autoimmunerkrankungen wie der systemische Lupus erythematodes zu nennen, denen eine verstärkte Aktivität von Interferonen gemeinsam ist. Interessanterweise verneinte der Patient, zeitlebens jemals die Diagnose oder Klinik eines viralen Infektes gehabt zu haben.
Eine erste human-genetische Vorstellung erfolgte im Alter von 33 Jahren und führte trotz wiederkehrender Vorstellungen bis zum 47. Lebensjahr nicht zur Identifikation der zugrunde liegenden Ursache der Beschwerden.
Die weiteren Jahre erbrachten keine neuen Ergebnisse, bis die Tochter des Patienten im Alter von 4 Jahren über Gelenkschmerzen zu klagen begann. Auch sie kam in spezialisierte, zahnmedizinische Betreuung, da Milchzähne nicht ausfielen und die bleibenden Zähne nicht durchbrachen. Im Rahmen einer Vorstellung in der Kinderrheumatologie der Charité im Alter von 8 Jahren zeigten sich in der Familienanamnese Parallelen zwischen Tochter, Vater und der Mutter des Vaters. Laborchemisch fiel erst bei der Tochter und in einer nachfolgenden Untersuchung auch beim Vater eine richtungsweisende, stark erhöhte Expression von SIGLEC1 auf. Es handelt sich hierbei um einen durch Typ-1-Interferon induzierten Oberflächenmarker auf Blutmonozyten. Die erneute Vorstellung der Tochter und des Vaters in der Humangenetik erbrachte nach weiteren Recherchen den konkreten Verdacht auf eine erbliche Interferonopathie, das Singleton-Merten-Syndrom. Durch eine daraufhin veranlasste gezielte Sanger-Sequenzierung (Verfahren zur Bestimmung der Reihenfolge der für ein Gen kodierenden Nukleotidbasen) des bei der Verdachtsdiagnose betroffenen IFIH1-Gens konnte eine neue Mutation nachgewiesen und so das Singleton-Merten-Syndrom genetisch gesichert werden [1].