Der systemische Lupus erythematodes (SLE) ist eine prototypische Autoimmunerkrankung, die überwiegend junge Frauen betrifft und praktisch alle Organe des Körpers schädigen kann. Obwohl wir während der letzten Jahrzehnte viele neue Einsichten in die Pathogenese des SLE gewonnen haben, ist unser Wissen trotzdem noch immer unvollständig. Neben einer genetischen Prädisposition spielen auch Umweltfaktoren, wie z. B. Zigarettenrauchen, Sonnenbrand, Infektionen oder Vitamin D-Mangel, eine entscheidende Rolle, sowohl bei der Krankheitsentstehung, als auch bei der Schubauslösung. Zahlreiche Manifestationen werden durch pathogene Autoantikörper hervorgerufen; somit sind B-Zellen und Plasmazellen an der Pathogenese unmittelbar beteiligt. Wichtige Erkenntnisse zu Ätiologie und Pathogenese sind in dieser Übersicht kurz zusammengefasst.
Schlüsselwörter: Systemischer Lupus erythematodes, Pathogenese, Umweltfaktoren, Immunkomplexe
Einführung
Der systemische Lupus erythematodes (SLE) gehört zur Krankheitsgruppe der Kollagenosen, entzündlichen Autoimmunopathien des Bindegewebes. Der SLE kann sich an praktisch allen Organen manifestieren. Am auffälligsten sind die Hauterscheinungen, besonders das schmetterlingsförmige Gesichtsexanthem. Arthralgien, oft auch eine Arthritis, kommen ebenfalls häufig vor. Organbedrohend ist die bei ca. 60% der SLE-Patienten auftretende Nierenbeteiligung. Herz- und ZNS-Manifestationen sind seltener, aber lebensgefährlich. In dieser kurzen Übersichtsarbeit kann nur auf einige wichtige Aspekte der Ätiologie und Pathogenese eingegangen werden, besonders aber auf solche Pathomechanismen, die (potenzielle) therapeutische Angriffspunkte darstellen.
Genetische Prädisposition
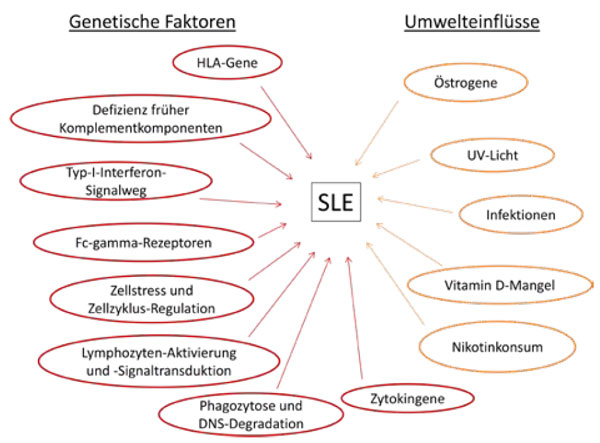
Die Bedeutung der genetischen Prädisposition geht klar aus Zwillingsstudien hervor. Wenn ein eineiiger Zwilling einen SLE hat, so hat der zweite Zwilling ein ca. 50-prozentiges Risiko, ebenfalls einen SLE zu entwickeln [1]. Derzeit sind ca. 100 SLE-assoziierte Genloci bekannt. Bei Erwachsenen findet sich fast immer ein polygener Vererbungsmodus, wobei neben SLE-spezifischen Risikoallelen auch solche eine Rolle spielen, die allgemein zu Autoimmunkrankheiten disponieren [2–5]. Hingegen finden sich bei sehr früher SLE-Manifestation im Kindesalter gelegentlich auch monogenetische Ursachen, z. B. eine C1q-Defizienz. Eine starke Assoziation mit dem SLE besitzen die MHC-II Gene HLA-DR2 und -DR3, welche Peptidantigene an T-Helferzellen präsentieren. Auch BLNK und PTPN22 sind in die Aktivierung des adaptiven Immunsystems involviert [5]. Weitere Risikogene sind Teil des Typ-I-Interferon-Systems, des NF-κB-Signalwegs, der Phagozytosemechanismen, der DNA-Abbauwege, Apoptoseregulation etc. [4, 5] (Abb. 1).
Umweltfaktoren
Umweltfaktoren besitzen einen vergleichbar starken Einfluss auf die Manifestation eines SLE wie genetische Risikofaktoren. Übermäßige UV-Bestrahlung, v. a. ein Sonnenbrand, induziert Apoptose und Nekrose in Hautzellen. Normalerweise phagozytieren spezialisierte Makrophagen rasch apoptotische Zellen, bevor sie zerfallen und Zellkernbestandteile einschließlich Nukleosomen – zentrale Autoantigene beim SLE – freisetzen.
Eine ausgeprägte Vitamin-D-Defizienz dürfte die Manifestation eines SLE begünstigen [6]. Da SLE-Patienten UV-Licht meiden sollen, verstärkt sich ein etwaiger Vitamin-D-Mangel weiter. Eine großzügige Vitamin-D-Substitution ist daher bei SLE-Patienten wichtig und dürfte sogar Krankheitsaktivität und Prednisolon-Bedarf reduzieren, möglicherweise u. a. durch die Induktion regulatorischer T-Zellen. Zigarettenrauchen, das zahlreiche entzündlich-rheumatische Krankheiten „befeuert“, begünstigt auch die Manifestation und Aktivität des SLE und reduziert die Wirkung der zur Behandlung wichtigen Antimalariamittel [7, 8].
Östrogene sind in die SLE-Pathogenese involviert, was zumindest teilweise erklärt, warum Frauen in der Zeit zwischen Menarche und Menopause etwa zehnmal häufiger erkranken als Männer. Dementsprechend können höher dosierte Östrogenpräparate, vor allem wenn sie keinen Gestagenanteil beinhalten, die Manifestation und Schübe eines SLE begünstigen. Moderne triphasische Antikonzeptiva mit niedrig dosiertem Östrogenanteil sind jedoch während Phasen niedriger oder stabiler Krankheitsaktivität ohne wesentliches Schubrisiko [9]. Östrogenhaltige Medikamente sollten jedoch bei Thrombosegefährdung, z. B. bei Nachweis von Antiphospholipid-Antikörpern oder Lupus-Antikoagulans, vermieden werden und können z. B. durch reine Gestagenpräparate (sog. Minipille) ersetzt werden.
Auch Infektionen, z. B. die durch das Epstein-Barr-Virus, kommen als Auslöser des SLE oder eines Krankheitsschubs in Frage. Hauptmechanismus sind vermutlich der durch Infektionen induzierte apoptotische Zelltod bei gleichzeitiger Aktivierung des Immunsystems, in bestimmten Fällen evtl. auch molecular mimicry sowie die Bildung von neutrophil extracellular traps (NETs). Abb. 1 fasst wichtige genetische Prädispositionen und Umweltfaktoren zusammen.