Trillium Diagnostik 2018; 16(4): 240-242
Unter den Strömungsbedingungen der Blutbahn ist eine gute Verankerung von Vorteil. Eine feste chemische Bindung hilft, Zellen oder Proteine an Ort und Stelle zu halten und anzureichern, um so lokal wichtige Funktionen ausführen zu können. Solche Vernetzungen knüpft die Transglutaminase Faktor XIII und spielt dadurch eine wichtige Rolle im Bereich der Wundheilung und Hämostase.
Schlüsselwörter: Immunturbidimetrie, Ammoniakfreisetzung, Isopeptidase-Aktivität, Gerinnsellöslichkeitstest, V34
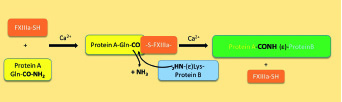
Üblicherweise sind aktivierte Gerinnungsfaktoren Serinproteasen oder deren Kofaktoren. Ihre Aufgabe besteht in der Spaltung von anderen Proteinen, meist im Sinne einer Aktivierung oder Inaktivierung. Ganz anders wirkt Faktor XIII (FXIII): Er gehört zu den Transglutaminasen, die im Körper Proteine und auch Zellen chemisch miteinander vernetzen können. In Plasma findet man FXIII, ein Protein mit ca. 320 kDA, als Heterotetramer A2B2 an Fibrinogen als Trägerprotein gebunden. Die A-Untereinheiten mit einem Cystein im aktiven Zentrum werden in Megakaryozyten und Makrophagen synthetisiert, die B-Untereinheiten in der Leber. Im Plasma bildet sich ein A2B2-Komplex, während in der Cerebrospinalflüssigkeit ein großer Teil der A-Untereinheiten in freier Form vorliegt [1]. In Megakaryozyten bzw. Plättchen, Chondrozyten, Osteoblasten, Osteozyten und in der Plazenta findet man die A2-Form. Die B-Untereinheiten schützen die A-Form vor vorzeitiger Proteolyse und Aktivierung, sind aber auch für die Substratbindung und die Aktivierung durch Thrombin wichtig. FXIII hat im Blut eine Halbwertszeit von etwa 9–14 Tagen; die Plasmakonzentration beträgt ca. 22 μg/ml [2]. Das Proenzym FXIII wird bei Gerinnungsaktivierung durch Thrombin und Kalziumionen in die aktive Transglutaminase FXIIIa aktiviert. Dabei wird je ein Aktivierungspeptid aus den A-Ketten abgespalten, anschließend lösen sich unter Mitwirkung von Ca2+-Ionen und Fibrin die B-Ketten ab. Bei einer der vielen genetischen Varianten, FXIII V34L, ist die Aktivierung beschleunigt. Auch die Komplementprotease MASP-1 kann FXIII (und andere Hämostaseproteine) aktivieren [3], und ist so ein Beispiel für die vielen Vernetzungen von Hämostase und Komplementsystem.