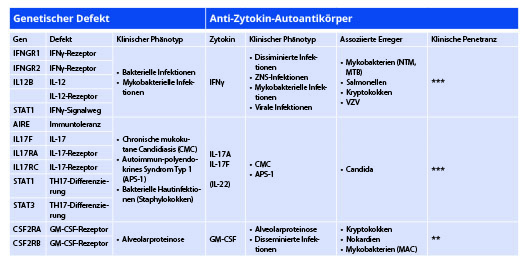
Autoantikörper gegen Zytokine werden bei ähnlicher klinischer Präsentation als Phänokopie angeborener Immundefekte angesehen. Sie prädisponieren zu Infektionen durch atypische Mykobakterien (Anti-IFNγ-Autoantikörper), verursachen mukokutane Candidiasis (Anti-IL-17A/F-Autoantikörper) oder pulmonale Alveolarproteinose mit oder ohne Anfälligkeit für Pilzinfektionen (Anti-GM-CSF-Autoantikörper) und können pyogene Infektionen (Anti-IL-6-Autoantikörper) begünstigen. Nicht alle Individuen mit Autoantikörpern gegen Zytokine erkranken an Infektionen, und nicht alle, die erkranken, erkranken gleich schwer. Die klinische Penetranz der Autoantikörper gegen Zytokine ist also variabel. Ob Autoantikörper gegen Zytokine krankheitsverursachend, also klinisch relevant sind, hängt von ihrer Konzentration und inhibierenden Wirkung auf die spezifische Zytokinfunktion ab. Da niedrig-titrige Autoantikörper gegen Zytokine häufig sind, kann die Diagnose eines „klinisch relevanten“, d. h. krankheitsverursachenden Anti-Zytokin-Autoantikörpers, erst nach Bestimmung des Antikörpertiters sowie dessen stark neutralisierender Wirkung auf die spezifische Zytokinfunktion gestellt werden.
Schlüsselwörter: Autoantikörper, Zytokin, Phänokopie, angeborener Immundefekt
Noch vor zehn Jahren waren vor allem angeborene, sogenannte primäre Immundefekte mit Abweichungen in Immunglobulinspiegeln und Zusammensetzungen der Lymphozytenpopulationen bekannt. Zwischenzeitlich sind mehr als 380 primäre Immundefekte beschrieben, die in neun verschiedene Gruppen unterteilt werden. Acht beschreiben angeborene Defekte im engeren Sinne, die durch Mutationen in der Keimbahn des Patienten verursacht werden: 1) Defekte der zellulären und humoralen Immunität, 2) Kombinierte Defekte der zellulären und humoralen Immunität mit assoziierten oder syndromalen Merkmalen, 3) Antikörpermangeldefekte, 4) Immundysregulationen, 5) Phagozytendefekte, 6) Defekte der intrinsischen und angeborenen Immunität, 7) Autoinflammationserkrankungen und 8) Komplementdefekte. Die neunte Gruppe umfasst Phänokopien angeborener Immundefekte [1]. Phänokopien sind Erkrankungen, deren klinische Präsentation angeborenen Immundefekten stark ähnelt, die aber nicht durch Mutationen in der Keimbahn hervorgerufen werden. Bisher bekannte Ursachen für Phänokopien angeborener Immundefekte sind entweder somatische Mutationen oder Autoantikörper gegen Zytokine und andere lösliche Proteine des Immunsystems. Die Beantwortung von drei Fragen erlaubt eine Einschätzung der biologischen Rolle von Autoantikörpern gegen Zytokine: a) Gibt es einen angeborenen Immundefekt mit Beeinträchtigung des korrespondierenden Zytokinrezeptors oder des korrespondierenden Zytokins mit ähnlicher klinischer Präsentation? b) Sind zahlreiche Patienten mit Autoantikörpern gegen Zytokine beschrieben worden, die an den gleichen schweren Erkrankungen wie Patienten mit korrespondierenden angeborenen Defekten leiden? c) Wie stark ist die klinische Penetranz? Die klinische Penetranz eines Autoantikörpers gegen Zytokine beschreibt dabei, wie viele Individuen mit Autoantikörpern gegen Zytokine auch erkranken und wie schwer diese Erkrankungen ausfallen. Die nachfolgende Übersicht wird den aktuellen Wissensstand für die vier wichtigsten bekannten Autoantikörper gegen Zytokine diskutieren.
Autoantikörper gegen IFNγ prädisponieren zu Infektionen durch nicht-tuberkulöse Mykobakterien, nicht-typhoidale Salmonellen und Varizellen
Erstmalig wurden neutralisierende Autoantikörper gegen IFNγ in einer Patientin thailändischer Herkunft mit tödlichen Infektionen durch Burkholderia cocovenenans und Mycobacterium cheloneae beschrieben [2]. Zwischenzeitlich sind mehr als 100 Patienten mit Autoantikörpern gegen IFNγ publiziert, die die Anfälligkeit dieser Patienten für schwere Infektionen durch nicht-tuberkulöse Mykobakterien (NTM) bestätigten, aber auch das Krankheitsspektrum dieser Patienten um schwere Infektionen durch nicht-typhoidale Salmonellen, Cryptococcus neoformans, Histoplasma capsulatum, Penicilium marneffi, Toxoplasma gondii und Varizella-Zoster-Viren erweitert haben [3, 4]. Autoantikörper gegen IFNγ wurden zunächst vor allem in Patienten südostasiatischer Herkunft identifiziert. Dieses Phänomen wird mit einer ethnisch spezifischen, HLA-restringierten Prädisposition für die Bildung von Antikörpern gegen Aspergillen, die mit IFNγ kreuzreagieren, erklärt [5]. Zwischenzeitlich wurde die Bildung neutralisierender Autoantikörper gegen IFNγ aber auch in einzelnen Patienten kaukasischer Abstammung beschrieben [3]. Untersuchungen zur Prävalenz von Autoantikörpern gegen IFNγ in verschiedenen Populationen wurden nicht durchgeführt, sodass eine genaue Abschätzung der klinischen Penetranz schwierig ist. Allerdings ähneln sowohl Erregerspektrum als auch die Schwere der Infektionen denen von Patienten mit angeborenen Defekten der Antwort auf oder Bildung von IFNγ [6]. Autoantikörper gegen IFNγ können daher mit hoher Sicherheit als Phänokopien dieser Immundefekte mit hoher klinischer Penetranz angesehen werden.