Schlüsselwörter: SCID, Immunphänotypen, Genetik, Stammzelltransplantation, Neugeborenenscreening
Einleitung
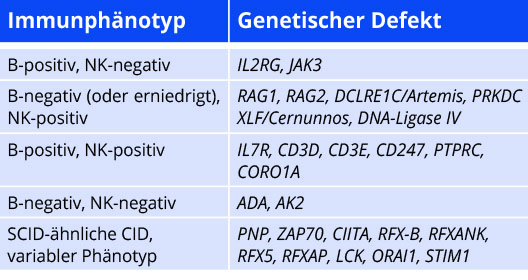
Schwere kombinierte Immundefekte (SCID für „Severe Combined Immunodeficiency“) sind eine Gruppe angeborener Immundefekte, die durch eine schwere Beeinträchtigung der zellulären und humoralen spezifischen Immunfunktionen definiert ist. Klinisch manifestiert sich die Erkrankung bereits im Säuglingsalter mit lebensbedrohlichen Infektionen. Kinder mit SCID versterben ohne Behandlung in der Regel vor dem zweiten Lebensjahr. Der Übergang von einem „schweren kombinierten Immundefekt“ (SCID) zu einem „kombinierten Immundefekt“ (CID) ist fließend. Die Zuordnung ist auch unter der Berücksichtigung klinischer, immunphänotypischer und genetischer Befunde in den ersten Lebensmonaten nicht für alle Patienten eindeutig möglich.
Klinische Präsentation
Infektionen
Die klinischen Manifestationen der Patienten mit SCID sind dominiert durch rezidivierende, ungewöhnlich schwere und somit atypische Verläufe von (opportunistischen) Infektionen. Beginn dieser Infektionen ist meist in den ersten Lebensmonaten. Betroffen sind häufig die Atemwege und der Gastrointestinaltrakt [1]. Typische Erreger sind Pneumocystis jirovecii, Adenovirus, Cytomegalievirus (CMV) oder das Respiratorische Synzytial-Virus (RSV). Eine Sonderrolle spielen Infektionen durch Lebendimpfungen: Kinder mit SCID fallen nach der seit 2013 von der STIKO empfohlenen Rotavirus-Lebendimpfung durch eine prolongierte Diarrhö mit Nachweis des Impfvirus auf. Dies ist ein klinisches Warnsignal, dem unbedingt nachgegangen werden sollte [2]. Auch andere Lebendimpfungen wie BCG oder die Polioschluckimpfung, welche in anderen Ländern regelhaft durchgeführt werden, können bei diesen Kindern zu lebensbedrohlichen Komplikationen führen.
Eine Sonderstellung nimmt die Retikuläre Dysgenesie ein: Neben dem Fehlen der Lymphozyten besteht eine Agranulozytose, sodass diese Kinder meist schon in den ersten Lebenstagen mit schweren bakteriellen Infektionen auffallen [3].
Maternofetale Transfusion
Maternale T-Zellen, die intrauterin an den Feten übertragen werden, können bei Fehlen der spezifischen zellulären Immunität nicht abgestoßen werden und persistieren im kindlichen Organismus. Hier treffen sie auf Alloantigene und können ein Bild verursachen, das klinisch dem einer Graft-versus-Host-Disease (GvHD) entspricht. Wie bei dieser können neben der Haut (Erythem) auch Darm (Enteritis) und Leber (cholestatische Hepatitis) beteiligt sein [4]. Diese Zeichen müssen sich nicht von Geburt an manifestieren und können sich auch erst in den ersten Lebensmonaten entwickeln. Die Differenzialdiagnose der Hauterscheinungen zu einem atopischen Ekzem oder Erythrodermien mit unterschiedlicher Ätiologie ist nicht immer einfach. Wichtig ist, bei diesen frühkindlichen Manifestationen an einen SCID zu denken und weitere diagnostische Schritte (s. unten) einzuleiten [4].
Als Ausdruck einer oligoklonalen peripheren Proliferation maternaler T-Zellen dominieren in der Immunphänotypisierung Gedächtnis-Zellen mit einem häufig eingeschränkten TCR-Repertoire [4]. Die maternalen T-Zellen können die Diagnostik erschweren: Expandierte maternale Zellen können zu normalen oder sogar erhöhten T-Zell-Zahlen im peripheren Blut führen, die HLA-Typisierung und genetische Diagnostik aus peripherem Blut kann durch die zusätzlichen mütterlichen Allele kompliziert werden. Bei Nachweis oder Verdacht auf eine MFT sollten genetische Untersuchungen und HLA-Typisierung nicht aus Vollblut oder Lymphozyten, sondern z. B. aus Granulozyten des peripheren Blutes durchgeführt werden.
Autoimmunität
In seltenen Fällen können autologe T-Zellen den klinischen Phänotyp eines SCID-Patienten wesentlich beeinflussen und die Diagnose erschweren. Auch kann eine ausgeprägte Leukozytose bestehen und den primären Verdacht in die Richtung einer leukämischen Erkrankung lenken. Der Nachweis autologer T-Zellen gelingt insbesondere bei Patienten mit hypomorphen Mutationen in Genen, die bei Nullmutationen einen „klassischen“ SCID-Phänotyp verursachen. Initial wurde dies bei Mutationen in den Recombinase Activating Genen (RAG1 oder RAG2) beobachtet, ist aber auch bei anderen Gendefekten beschrieben [5, 6]. Durch die Restfunktion der durch die hypomorphe Mutation veränderten Proteine, können einige T-Zellen ausreifen, treffen in ihrer Entwicklung jedoch auf eine insuffizient ausgebildete Thymusarchitektur, die keine zuverlässige Negativselektion autoreaktiver T-Zellen erlaubt. Diese wenigen autologen T-Zellen können bei Kontakt zu ihren Antigenen aktiviert werden und oligoklonal expandieren. Durch diese Autoimmunreaktionen können z. T. schwerste Organstörungen verursacht werden.
Diese besondere Manifestation des SCID durch Expansion autologer T-Zellen wird nach ihrem Erstbeschreiber als Omenn-Syndrom (OS) bezeichnet [7]. Patienten mit OS fallen durch Neurodermitis-ähnliche Exantheme, Alopezie, Kolitis bzw. Hepatitis ohne Erregernachweis auf, sowie durch Lymphadenopathien und Splenomegalie. Im Blutbild finden sich häufig eine Eosinophilie und im Serum z. T. deutlich erhöhte Spiegel für IgE. T-zelluläre Infiltrate mit eingeschränktem T-Zellrezeptor-Repertoire können sich in unterschiedlichen Geweben befinden, nicht unähnlich der GvH-Manifestation bei MFT (s. oben). Patienten jenseits des Säuglingsalters mit hypomorphen RAG-Defekten können durch Granulome an Haut, Schleimhaut und inneren Organen auffallen. Die histologisch epitheloidzelligen Granulome weisen T-zelluläre Infiltrate auf und dürfen nicht mit leukämischen Infiltraten verwechselt werden. Sie manifestieren sich z. B. an der Haut sowohl als papulöse als auch ulzerierende Effloreszenzen [8, 9]. Aus manchen dieser Granulome konnte Rötelnvirus isoliert werden, sodass ein infektiologischer Trigger postuliert wird [10].