Michael C. Frühwald, Johannes Holzapfel, Martin Hasselblatt
Als Entität sind ZNS-Tumoren die häufigsten soliden Neoplasien des Kindes- und Jugendalters. Bis zu 500 Betroffene werden jährlich an die Kompetenzzentren des Deutschen HIT-Netzwerkes (HIrnTumoren) gemeldet. Unter diesen machen die niedriggradigen Gliome annähernd 50% aus. Häufigste bösartige ZNS-Tumoren bei Kindern sind die Medulloblastome. Während die neuroradiologische und -pathologische sowie die Liquor-Diagnostik des HIT-Netzwerkes seit Jahrzehnten in Referenzzentren etabliert sind, nehmen neuere Methoden, wie der Methylierungs-Classifier zur genaueren Einstufung der ZNS-Tumoren, an Bedeutung zu.
In bestimmten Risikokonstellationen kann mit konventionellen Verfahren wie Neurochirurgie, adjuvanter Chemotherapie sowie ggf. Hochdosis-Chemotherapie und Strahlentherapie ein großer Anteil der Kinder und Jugendlichen geheilt werden. Neue Verfahren mittels zielgerichteter Substanzen werden jedoch dringend benötigt, um auch bei Hochrisiko-Neoplasien, wie den malignen Mittellinien-Gliomen, seltenen embryonalen ZNS-Tumoren wie ATRT und ETMR, aber auch metastasierten Erkrankungen bei Säuglingen und Kleinkindern, Fortschritte zu ermöglichen.
Ein besonderer Fokus der Betreuung von Patienten mit ZNS-Tumoren liegt auf der frühzeitigen Erkennung und adäquaten Behandlung von Spätfolgen der Tumorerkrankung, aber auch der tumororientierten Therapie.
Schlüsselwörter: ZNS-Tumoren, HIT-Netzwerk, Methylierungs-Classifier, zielgerichtete Therapien, Spätfolgen
Epidemiologie von ZNS-Tumoren im Kindes- und Jugendalter
Tumoren des ZNS sind bei deutschen Kindern und Jugendlichen die häufigsten soliden Neoplasien [1]. 2009–2013 wurden im Durchschnitt jährlich 480 Kinder und Jugendliche mit ZNS-Tumoren in die Studien des Behandlungsnetzwerks HIT gemeldet (A. K. Gnekow, Erfassung 2018). Sie treten vermehrt bei Jungen auf (Verhältnis männlich : weiblich 1,2 : 1) und neigen zu einer Lokalisation in der hinteren Schädelgrube. Bei Patienten unter zwei Jahren ist die supratentorielle Region mit bis zu 60% die häufigste Lokalisation [2].
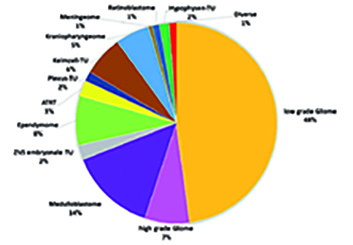
Histopathologisch finden sich unter den ZNS-Tumoren bei Kindern und Jugendlichen v. a. niedriggradige Gliome (Low-grade glioma, LGG: 45–50%), gefolgt von Medulloblastomen (ca. 20%), Ependymomen (ca. 10%), Kraniopharyngeomen (ca. 5%) und Keimzelltumoren (ca. 5%; Abb. 1; [2]). Seltenere Entitäten (1–3%) sind Tumoren des Plexus chorioideus (Plexus-Papillome und -karzinome), die atypischen teratoiden/rhabdoiden Tumoren (ATRT) sowie andere seltene embryonale Tumoren, wie die erst kürzlich molekular definierten embryonalen Tumoren mit mehrschichtigen Rosetten (ETMR) und die Pinealis-Tumoren [3].
Die prozentuale Verteilung der verschiedenen Entitäten variiert stark mit dem Alter. So sind ATRT bei Kindern in den ersten sechs Monaten die häufigsten bösartigen ZNS-Tumoren [4]. Zudem nimmt mit steigendem Alter die Inzidenz an Ependymomen, Medulloblastomen und anderen embryonalen ZNS-Tumoren ab. LGG zeigen im Kindesalter einen mehrgipfligen Verlauf mit Höhepunkten um das zweite, vierte bis siebte und zwölfte Lebensjahr [5].
Eine Vielzahl neuer Publikationen zu hochauflösenden molekulargenetischen Untersuchungen hat dazu beigetragen, ZNS-Tumoren weiter zu differenzieren. Insbesondere mithilfe von Methylierungs-Mustern lassen sich viele histomorphologisch kaum unterscheidbare Entitäten in Untergruppen einteilen [6]. Therapieansätze gerade bei Hochrisiko-Neoplasien, wie z. B. den diffus intrinsischen Pons-Gliomen (DIPG) oder auch den H3K27-positiven Mittellinien-Gliomen, orientieren sich nun oft primär an den zugrunde liegenden molekulargenetischen Veränderungen und nicht an der Histomorphologie.