T-Lymphozyten erkennen Antigen, das ihnen von antigenpräsentierenden Zellen dargeboten wird
Die Geschichte, die es hier zu erzählen gilt, hat zwei Kapitel. Das erste beginnt Ende der 1980er-Jahre. Damals wusste man, dass eine Gruppe von weißen Blutkörperchen, die T-Lymphozyten, von zentraler Bedeutung für die Abwehr von Infektionskrankheiten und die Regulation von Immunantworten ist. Grundsätzlich war auch bekannt, wie T-Lymphozyten aktiviert werden. Eine Besonderheit von T-Lymphozyten ist, dass sie ausschließlich Peptide, also Bruchstücke von Eiweißen, erkennen. Andere Substanzklassen, z. B. Zucker oder Fette entgehen also der Aufmerksamkeit der T-Lymphozyten. Erschwert wird die Aktivierung von T-Lymphozyten weiter dadurch, dass sie die Peptide nicht etwa in freier Form erkennen, sondern ausschließlich dann, wenn ihnen diese Peptide von körpereigenen Zellen präsentiert werden. Zur Peptidpräsentation dienen sogenannte HLA (human leukocyte antigen)-Moleküle. Die Mitte der 1970er-Jahre in Maus-Experimenten gemachte Entdeckung, dass T-Lymphozyten ausschließlich Peptide erkennen, die ihnen von körpereigenen HLA-Molekülen präsentiert werden, wurde 1996 mit dem Nobelpreis für Rolf Zinkernagel und Peter Doherty ausgezeichnet.
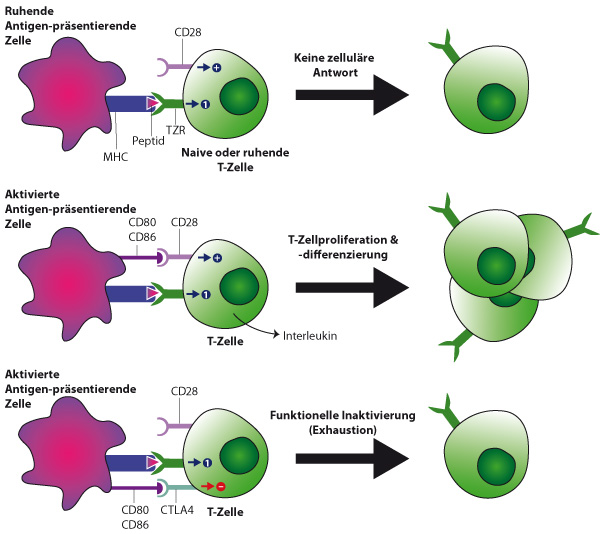
Die antigenpräsentierenden Zellen müssen den T-Zellen ein ko-stimulatorisches Signal geben, Antigenerkennung alleine reicht nicht zur T-Zellaktivierung
Mitte der 1980er-Jahre wollte Marc Jenkins im Labor von Ron Schwartz genauer wissen, wie die Präsentation von Peptid-Antigenen für T-Lymphozyten funktioniert. Er immunisierte Mäuse mit verschiedenen Antigenen und untersuchte die Aktivierung von T-Zellen, die aus diesen Mäusen gewonnen wurden. Dabei stellte sich heraus, dass die Aktivierung von T-Zellen nur dann gelang, wenn die antigenpräsentierenden Zellen vital waren. Chemisch inaktivierte antigenpräsentierende Zellen induzierten zwar die Aktivierung intrazellulärer Signalwege durch den T-Zellrezeptor, die T-Zellen reagierten trotzdem nicht. Im Gegenteil: T-Zellen, denen ihr spezifisches Antigen von chemisch inaktivierten antigenpräsentierenden Zellen dargeboten wurde, konnten später auch dann nicht mehr aktiviert werden, wenn ihnen das Antigen von intakten antigenpräsentierenden Zellen dargeboten wurde [1]. Die T-Zellen waren anerg, also gewissermaßen stummgeschaltet worden. In weiteren Experimenten konnten Jenkins und Schwartz zeigen, dass die Zugabe von sogenannten allogenen antigenpräsentierenden Zellen, die selber nicht in der Lage waren den T-Zellen das Antigen zu präsentieren, die Inaktivierung der T-Zellen durch die chemisch inaktivierten antigenpräsentierenden Zellen verhindern konnte. Zusammengefasst bedeuten diese Ergebnisse also: Die bloße Erkennung von Antigen durch den T-Zellrezeptor bewirkt keine Aktivierung der T-Zelle. Die antigenpräsentierende Zelle muss zusätzliche aktivierende Signale an die T-Zelle senden. Damit war die Suche nach diesen so wichtigen ko-stimulatorischen Signalen eröffnet.
CD28 ist ein ko-stimulatorischer Rezeptor auf T-Zellen und B7 ein ko-stimulatorischer Ligand auf antigenpräsentierenden Zellen
Ebenfalls Mitte der 1980er-Jahre hatte eine Arbeitsgruppe um Jeffrey Ledbetter Mäuse mit humanen T-Zellen immunisiert und danach Antikörper aus diesen Mäusen gewonnen, die unterschiedliche Oberflächenmoleküle der humanen T-Lymphozyten erkannten. Einer dieser Antikörper war spezifisch für ein Molekül, das heute als CD28 bekannt ist. Ledbetter und Kollegen berichteten, dass die Stimulation von CD28 alleine keinerlei Reaktion der T-Zellen hervorruft. Allerdings konnten T-Zellantworten, die durch Stimulation des T-Zellrezeptors induziert wurden, durch die gleichzeitige Stimulation von CD28 massiv verstärkt werden [2]. CD28 ist also ein ko-stimulatorischer Rezeptor auf T-Zellen. 1990 entdeckten Linsley, Ledbetter und Kollegen dann, dass CD28 an ein Molekül bindet, das sogenannte B7, das auf antigenpräsentierenden Zellen präsentiert wird [3]. B7 ist also ein ko-stimulatorischer Ligand auf antigenpräsentierenden Zellen. Anfang der 1990er-Jahre war also klar, dass die Interaktion zwischen B7 auf den antigenpräsentierenden Zellen und CD28 auf den T-Zellen das ko-stimulatorische Signal vermittelt, das zusätzlich zur Antigenerkennung durch den T-Zellrezeptor notwendig ist, um T-Zellen zu aktivieren. Sofort war klar, dass die Manipulation der ko-stimulatorischen Interaktionen zwischen antigenpräsentierenden Zellen und T-Zellen neue therapeutische Perspektiven entwickeln würde. In der Tat ist die therapeutische Blockade von CD28 (Abatacept) heute ein erfolgreich eingesetztes Mittel in der Therapie von Autoimmunkrankheiten. Für die Entwicklung der Checkpoint-Inhibitoren zur Therapie von Tumorpatienten war eine weitere Serie von Entdeckungen entscheidend.