Der Beginn – 1890 in Berlin
Antikörper sind Eiweißmoleküle, die von sogenannten Plasmazellen zur Abwehr von pathogenen Keimen in das Gewebe abgegeben werden. Plasmazellen entstehen aus ruhenden B-Lymphozyten (kurz: B-Zellen) nach Aktivierung durch z. B. ein Toxin oder Pathogen. Die Erfolgsgeschichte der Antikörper beginnt mit der Beobachtung von Gameleia (1889), Bouchard (1890) und Behring & Nissen (Mai 1890), dass das Blut von Tieren erst nach der Infektion mit Bakterien eine zerstörende Wirkung auf Bakterien in Kulturen zeigte [1]. Der Siegeszug der Antikörper begann aber erst mit der am 4. Dezember 1890 publizierten und in allen Lehrbüchern als Geburtsstunde der Antikörper gepriesenen Entdeckung von Emil von Behring und Shibasaburo Kitasato. Beide Forscher demonstrierten, dass Tetanus-erkrankte Mäuse durch die Übertragung einer im Serum von Tetanus-immunen Kaninchen vorhandenen antitoxischen Aktivität geheilt werden können [2]. Obwohl in dieser Publikation im Titel auch die Immunität gegen Diphtherie erwähnt wurde („Ueber das Zustandekommen der Diphtherie-Immunität und der Tetanus-Immunität bei Thieren“), wurden nur Daten über die Serumtherapie von Tetanus-erkrankten Mäusen beschrieben. Die Erzeugung einer Diphterie-Immunität in Labortieren wurde alleine von Emil von Behring am 11. Dezember 1890 [3], und die erfolgreiche Serumtherapie von an Diphtherie erkrankten Meerschweinchen von Emil von Behring zusammen mit Erich Wernicke im Jahre 1892 [4] publiziert. Emil von Behring erhielt 1901 den ersten Nobelpreis für Physiologie und Medizin für „his work on serum therapy, especially its application against diphtheria“ 1.
Nach der erfolgreichen Anwendung der Serumtherapie im Tiermodell nahm die Umsetzung vom Labor in die Klinik, vor allem auch durch die Mitarbeit von Paul Ehrlich, eine für heutige Zeiten rasante Fahrt auf. Ehrlich gelang es, durch Verwendung von Ziegen und Pferden eine größere Menge an Seren zu produzieren und in diesen den Gehalt der antitoxischen Aktivität zu quantifizieren, d. h. den Titer der Antikörper gegen Diphtherie zu bestimmen [5].
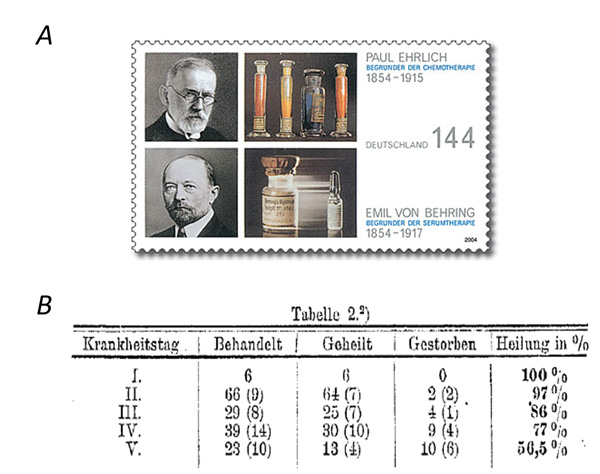
Diese standardisierten Anti-Diphtherie-Seren mit bekannten „Immunitätseinheiten“ wurden dann unter der Anleitung von Paul Ehrlich und Emil von Behring zum ersten Mal an insgesamt 220 an Diphtherie erkrankten Kindern in sechs Berliner Krankenhäuser auf ihre Wirksamkeit überprüft [6]. Das Ergebnis war phänomenal (Abb. 1): Während 50% der Kinder verstarben, die kein Antiserum erhielten und nur mit einem damals üblichen Luftröhrenschnitt behandelt wurden, konnten 64 von 66 Kindern, die früh nach dem Auftreten der Symptome (I. und II. Krankheitstag in Tabelle 1) mit dem Antiserum behandelt wurden, geheilt werden. Für die im Serum enthaltene antitoxische Aktivität setzte sich letztendlich der von Paul Ehrlich im Jahre 1891 geprägte Begriff „Antikörper“ durch [7].
Paul Ehrlich war auch der Erste, der in seiner Seitenkettentheorie ein bis heute in seinen Grundzügen gültiges Modell zur Erklärung der Bildung spezifischer und löslicher Antikörper gegen ein Toxin präsentierte (Abb. 2) [8]. Für diesen Beitrag erhielt er 1908 zusammen mit Ilya Metchnikoff, dem Entdecker der Bakterien-fressenden Phagozyten (und somit der ersten Immunzelle), den Nobelpreis für Medizin. Die Entdeckung von Almroth Wright, dass Immunseren gegen Bakterien diese im Zusammenspiel mit Phagozyten besser abtöteten [9], führte zu einem weiteren bis heute gültigen Paradigma in der Immunologie: „Infektionen werden effizienter durch das Zusammenwirken der zellulären und der humoralen Immunität bekämpft“.
Nachdem Karl Landsteiner in den 1920er-Jahren zeigen konnte, dass Antikörper auch gegen kleine organische Moleküle (Haptene) nach Kopplung an einen Proteinträger („Carrier“) gebildet wurden [10], kamen starke Zweifel an der Seitenkettentheorie auf, vor allem weil es schwer vorstellbar war, dass für so viele Seitenketten genügend Platz auf einer Zelle vorhanden ist. Ehrlichs Theorie geriet schließlich ganz in Vergessenheit. Erst 1957 wurde in der von Frank Macfarlane Burnet publizierten klonalen Selektionstheorie Ehrlichs Idee wieder aufgegriffen (Abb. 2), was auch gebührlich in der Publikation von Burnet erwähnt wurde [11].