Das Immunsystem wird im Allgemeinen unterteilt in das angeborene (innate) und das erworbene (adaptive) Immunsystem. Die Besonderheit des erworbenen Immunsystems besteht in der Fähigkeit, Rezeptoren, die exakt auf bestimmte Proteinstrukturen passen, im Lauf des Lebens neu zu bilden. Dies geschieht über den Mechanismus der Rekombination und klonalen Selektion in T- und B-Zellen, bei der verschiedene Gen-Abschnitte neu miteinander verknüpft werden. Dadurch entstehen immer wieder neue Proteine, die bestimmte Proteinstrukturen mit hoher Spezifität erkennen. Die resultierenden Rezeptoren sind die T-Zell-Rezeptoren (TCR) und die B-Zell-Rezeptoren (BCR) bzw. die dann sezernierten Antikörper.
Während dieser Teil des Immunsystems ein ausgeklügeltes System für die Erkennung von neuen Proteinstrukturen vorhält, mit denen der Körper noch nie vorher in Kontakt war, basiert das angeborene Immunsystem auf Rezeptoren, die bereits genetisch angelegt sind, und in der Regel hochkonservierte molekulare Strukturen eingedrungener Krankheitserreger erkennen. Ein Beispiel ist der Immunrezeptor TLR4 (Toll-like-Rezeptor 4), der das Lipopolysaccharid (LPS) in der Zellwand gramnegativer Bakterien detektiert. Neben TLR4 gibt es eine Reihe weiterer Mustererkennungsrezeptoren (pattern recognition receptors), die Pathogen-assoziierte molekulare Muster (pathogen-associated molecular patterns) erkennen. Sie werden zu Rezeptorfamilien zusammengefasst: z. B. den Toll-like-Rezeptoren, den NOD-like-Rezeptoren oder den RIG-I-like-Rezeptoren.
Grundzüge der Immunerkennung von Nukleinsäuren
Nukleinsäuren sind ganz grundsätzliche Bausteine allen Lebens, und ihre molekulare Grundstruktur ist von Bakterien bis hin zu Wirbeltieren universell. Eine Erkennung von fremder DNA und RNA kann daher nicht nach den Prinzipien der molekularen Mustererkennung erfolgen, wie beispielsweise bei fremden Molekülstrukturen wie LPS. Das Eindringen fremder Nukleinsäuren bedeutet jedoch für alle Lebensformen Gefahr. Das Abwehren dieser Gefahr ist daher eine der ureigenen Aufgaben des Immunsystems.
Die Biologie hat auf allen Stufen der Evolution Mechanismen zur Erkennung und Eliminierung fremder Nukleinsäuren etabliert, die heute in ihrer Gesamtheit als Nukleinsäure-Immunität bezeichnet werden [1].
Diese Mechanismen gehen über die eigentlichen Immunzellen wie beispielsweise Monozyten, Granulozyten, oder T-Zellen hinaus. Alle Körperzellen sind in der Lage, fremde Nukleinsäuren zu erkennen, und entsprechende Abwehrmechanismen einzuleiten. Dennoch sind bestimmte Immunzellen wie z. B. die plasmazytoiden dendritischen Zellen (pDC) auf die Erkennung spezialisiert und steuern die Sensitivität der Nukleinsäure-Immunerkennung in anderen Körperzellen.
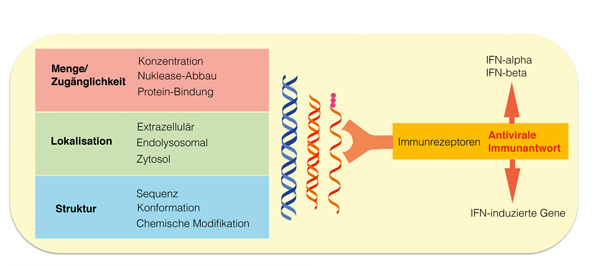
Für eine treffsichere Erkennung fremder Nukleinsäuren werden dreierlei Informationen integriert:
ihre Menge und die Zugänglichkeit für eine Erkennung,
bestimmte strukturelle Komponenten (Abb. 1).
Die Menge und Zugänglichkeit der Nukleinsäuren für eine Immunerkennung wird durch zwei Faktoren bestimmt. Ihre Konzentration in Verbindung mit der Nukleaseaktivität in der direkten zellulären Umgebung, und ihre Bindung an weitere Komponenten wie Proteine, die wiederum ihre Stabilität und Erkennbarkeit beeinflussen.
Die Lokalisation verschiedener Nukleinsäuren, z. B. außerhalb der Zelle, im endolysosomalen Kompartment der Zelle, im Zytosol oder im Zellkern, gibt Information darüber, ob ihr Vorkommen physiologisch oder ungewöhnlich ist.
Besondere strukturelle Merkmale, die über die grundsätzliche molekulare Struktur hinausgehen, tragen zur Erkennung von Nukleinsäuren bei. Dabei werden solche Merkmale einerseits von der Zelle als Signaturen für Selbst-Nukleinsäure in die Nukleinsäure eingebaut und dann als selbst detektiert; andererseits können bestimmte strukturelle Merkmale auf fremde Herkunft hinweisen. Beispiele hierfür sind lange Doppelstrang-RNA, oder DNA im Zytoplasma.
Spezialisierte Immunrezeptoren nutzen die drei Informationsarten zur Erkennung von fremden Nukleinsäuren. Diese führt dann zur Aktivierung der Rezeptoren und der nachfolgenden Signalwege. Die resultierende Immunantwort ist gegen eindringende Fremd-Nukleinsäuren gerichtet, also in der Regel Viren oder intrazelluläre Bakterien. Charakteristisch ist die Induktion von Typ-I-Interferon (IFN-alpha und IFN-beta) und einem breiten Spektrum von IFN-induzierbaren Genen. Weiterführende Informationen zur Nukleinsäure-Immunität sind in folgendem Übersichtsartikel aktuell zusammengefasst [1].