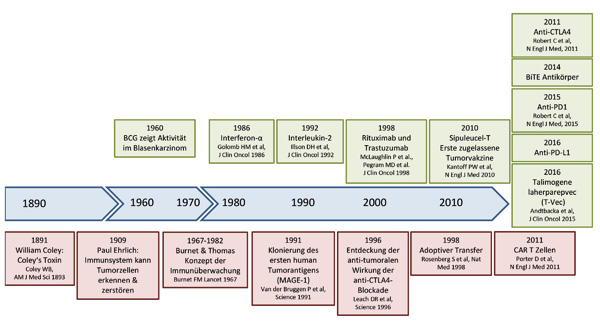
Meilensteine der Krebsimmuntherapie
Die Idee, dass unser Abwehrsystem prinzipiell in der Lage ist, entartete Zellen zu erkennen und zu zerstören, hat eine lange Historie. Bereits im alten Ägypten wurden Tumorregressionen nach Infektionskrankheiten beobachtet und daraufhin die Induktion einer Infektion zur Tumorbehandlung empfohlen, wie im Papyrus Ebers aus dem 16. Jahrhundert vor Christus festgehalten wurde. Der Bonner Chirurg Wilhelm Busch hat bereits 1866 ein klinisches Experiment publiziert, bei dem er durch die beabsichtigte Auslösung eines Erysipels im Tumorbereich bei einem Patienten die beinahe komplette Rückbildung des Tumors beobachtete. Später ging der amerikanische Arzt William Bradley Coley als Pionier der Krebsimmuntherapie in die Medizingeschichte ein. Dieser hatte 1891 durch die Injektion von Streptokokken in Sarkome die Regression von Tumoren erreicht. Bis zur Entdeckung der Chemo- und Strahlentherapie war die als Coley’s Toxin bezeichnete Mischung aus abgetöteten Bakterien die einzige systemische Krebstherapie. Das Wirkprinzip hat bis heute durch die Verwendung von Bacillus Calmette-Guérin (BCG) zur Behandlung des frühen Blasenkarzinoms Bestand. Paul Ehrlich formulierte 1909 die These, dass das Immunsystem Tumorzellen beseitigen kann und dadurch die Entstehung von Krebs verhindert. Lewis Thomas und Frank Macfarlane Burnet vermuteten ab den 1950er-Jahren, dass
T-Zellen eine wesentliche Rolle bei der immunologischen Überwachung spielen. Den Beweis, dass T-Zellen das Tumorantigen MAGEA1 (melanoma antigen family A,1) erkennen können, lieferte Pierre van der Bruggen 1991. Dass T-Zellen im Patienten zu Tumorremissionen führen können, wurde erstmalig 1998 durch adoptive T-Zell-Transfer-Studien in Kombination mit Interleukin-2 an metastasierten Melanompatienten von Steven Rosenberg gezeigt. 2011 hat die Aktivierung von T-Zellen durch Checkpoint-Inhibitoren, basierend auf den experimentellen Arbeiten von James Allison, zum Durchbruch der Therapie des metastasierten Melanoms geführt. Aktuell führen die Immun-Checkpoint-Inhibitoren bei einer zunehmenden Anzahl von Patienten mit unterschiedlichen, fortgeschrittenen Tumorerkrankungen zu langfristigen Remissionen. Noch am Anfang steht die Entwicklung von bispezifischen T-Zell-aktivierenden Antikörpern (sog. BITE = bispecific T-cell engager). Diese erkennen neben CD3 auf der T-Zelle ein zweites Antigen auf der Oberfläche der Tumorzellen, wodurch Tumor und T-Zelle direkt in Kontakt gebracht und die Lyse der Tumorzelle induziert wird. Eine weitere neue innovative T-Zell-gerichtete Therapie ist der adoptive T-Zell-Transfer mit chimären Antigenrezeptor (CAR)-transduzierten T-Zellen. Dabei werden T-Zellen aus dem peripheren Blut durch genetische Modifikationen mittels viralem Gentransfer moduliert. Nach dem Schlüssel-Schloss-Prinzip erkennen die sog. chimären Antigenrezeptoren ein spezielles Peptid auf der Tumorzelle, die T-Zelle wird aktiviert und lysiert die Antigen-tragende Zelle. Mithilfe der CRISPR/Cas-Technologie sollen T-Zellen von Patienten zukünftig so manipuliert werden können, dass z. B. ihre Effektorfunktion erhöht wird. Diese Erkenntnisse revolutionieren unser Verständnis der Tumorbiologie und Tumorimmunologie. Neben der Tumorzelle dient jetzt auch das Immunsystem als Zielstruktur für neue Therapie-Ansätze. Tumorerkrankungen, die bisher fast ausschließlich als genetische Erkrankungen der Tumorzelle selbst verstanden wurden, müssen nun auch als Erkrankung der Immunregulation verstanden werden.