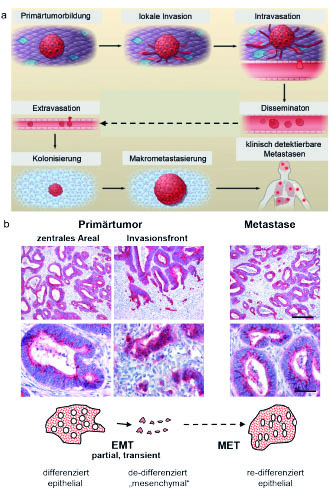
Metastasierung ist die Haupttodesursache bei soliden Tumoren. Neben der Akkumulation und Selektion von Mutationen ist die erhöhte phänotypische Plastizität von Krebszellen eine wichtige Triebkraft der Tumorprogression. Diese Plastizität erlaubt eine dauernde Anpassung der Tumorzelle an sich permanent ändernde äußere Bedingungen auf dem Weg vom Primärtumor bis zur Makrometastase. Dementsprechend kommt der Wechselwirkung zwischen Tumorzelle und Tumorumgebung, insbesondere mit Zellen des Immunsystems, eine zentrale Bedeutung für die Tumorprogression zu. Dabei ermöglicht die transiente und partielle Aktivierung des embryonalen Programms der epithelialen-mesenchymalen Transition (EMT), sowie daran gekoppelter Stammzell- und Überlebens-Eigenschaften, die effiziente Disseminierung widerstandsfähiger Tumorzellen. Die nachfolgende Rückdifferenzierung zum epithelialen Phänotyp (MET) fördert starkes Metastasenwachstum. Ein Verständnis der zugrunde liegenden intra- und extrazellulären molekularen Mechanismen ist die Basis neuer therapeutischer Konzepte gegen die tödliche Metastasierung.
Schlüsselwörter: Plastizität, EMT, Metastasierung, Tumorstammzellen, Tumorimmunologie
Von der Tumorentstehung zur Metastase – zelluläre Plastizität als Triebkraft
Für über 90 Prozent aller Krebstodesfälle bei soliden Tumoren (v. a. Karzinomen) ist nicht der Primärtumor verantwortlich. Die Patienten versterben an den Metastasen. Um Krebs besser behandeln zu können, muss man daher vor allem verstehen, wie Metastasen entstehen. Karzinome bilden sich aus ortsständigen, epithelialen Geweben. Wie schafft es ein aus immobilem Gewebe entstehender Tumor überhaupt, sich im Körper auszubreiten und Fernmetastasen zu bilden? Dazu müssen Krebszellen die Fähigkeit erwerben, sich vom Primärtumor abzulösen, sich über Blutgefäße im Körper zu verteilen (Dissemination) und in anderen Organen, z. B. in Leber und Lunge, wieder anzuwachsen (Kolonisierung) und sich dort zu vermehren (Mikro-, Makrometastasen) (Abb. 1a) [1, 2]. Zudem generieren Krebszellen Neoantigene und sind daher permanent potenziellen Angriffen des Immunsystems ausgesetzt. Ein hürdenreicher Weg, den glücklicherweise nur wenige Tumorzellen beenden können. Die zugrunde liegenden molekularen Mechanismen sind nicht vollständig verstanden. Die allgemein akzeptierte Tatsache ist, dass die Tumorprogression, von der Initiation, über gutartige Vorstufen bis zur finalen Metastasierung durch die schrittweise Akkumulation und Selektion von Mutationen in Onkogenen und Tumorsuppressorgenen getrieben wird. Dieser lineare Prozess – Mutationen sind irreversibel – kann aber nicht alle Beobachtungen im Zuge der Metastasenbildung erklären. Vor ca. 20 Jahren fiel uns auf, dass gut differenzierte Tumoren an der Invasionsfront de-differenzieren und separierte, kleine Tumorzell-Cluster oder Einzelzellen bilden, die somit potenziell leicht in Blutgefäße eindringen und disseminieren können. Erstaunlicherweise sind Metastasen solcher Tumoren meistens wieder gut differenziert [3] (Abb. 1b). Offenbar können Krebszellen transient ihren Phänotyp wechseln und sich damit an die sich laufend ändernden Bedingungen einer fremden Umgebung im Zuge der Metastasierung anpassen, was sich mit irreversiblen Mutationen nicht vereinbaren lässt. Die Grundlage hierfür – so die resultierende Hypothese – ist eine abnorme Plastizität der Tumorzellen, die neben der Akkumulation von Mutationen, bzw. auf deren Basis, als eine zweite zentrale Triebkraft der Progression bis zur Metastasierung fungiert. Was ist die Grundlage dieser Plastizität von (einigen) Tumorzellen innerhalb eines Tumors?