Schlüsselwörter: Immundefekt, B-Zellen, T-Zellen
Einleitung
Stellt man sich die Aktivität des menschlichen Immunsystems als Kontinuum vor, so erscheint es einfach: Ein überaktives Immunsystem sorgt zwar für einen guten Schutz vor Krankheitserregern und anderen äußeren Gefahren, attackiert jedoch den eigenen Körper. Diese Gefahr wird durch ein inaktiveres Immunsystem vermieden, jedoch zum Preis von vermehrten Infektionen. Hier erschiene wenig einleuchtend, wie Immundefizienz mit Autoimmunität einhergehen kann. Die Realität in der Klinik zeigt jedoch, dass dieses Modell zu einfach ist, denn Autoimmunität kann die Folge eines dysfunktionalen Immunsystems sein. Immundefizienz und Autoimmunität können somit, scheinbar paradoxerweise, zwei Seiten einer Medaille darstellen.
Die komplexen Wechselwirkungen zwischen Immundefizienz und Autoimmunität lassen sich einteilen in primäre, sekundäre und tertiäre Assoziationen. Diese Arbeit betrachtet in erster Linie das gemeinsame Auftreten von Immundefizienz und Autoimmunität im Rahmen von genetischen Erkrankungen, also die primären Assoziationen. Sekundär kann eine Manifestation eine andere verursachen, so wie beispielsweise eine autoimmune Zerstörung von neutrophilen Granulozyten zu Immundefizienz führt. Schließlich kann in tertiären Fällen die Immundefizienz eine Nebenwirkung der Behandlung der Autoimmunität sein.
Genetische Risikofaktoren
Primäre Immundefekte und rheumatische Erkrankungen zählen zu den genetisch am intensivsten erforschten immunologischen Erkrankungen. So konnten durch eine Metaanalyse Genom-weiter Assoziationsstudien mit fast 30.000 an rheumatoider Arthritis erkrankten Patienten und nachfolgende bioinformatische Analyse 98 Gene (außerhalb der bekannterweise für die Krankheitsentstehung wichtigen Haupthistokompatibilitätskomplexe (MHC)) identifiziert werden, welche mit der Erkrankung assoziiert waren [1]. Durch Varianten in den Genen wurde das Erkrankungsrisiko bei dieser polygen vererbten Krankheitsform mindestens verdoppelt. In 15 dieser 98 Gene sind kausale Varianten für monogen vererbte primäre Immundefektsyndrome bekannt [2,3]. Auch für den systemischen Lupus erythematodes sind über 50 Risikoloci bekannt, welche außerhalb der MHC-Regionen liegen. Hierzu zählen Varianten, welche den Interferon-alpha-Stoffwechselweg beeinflussen, und Gene aus dem Komplementsystem. Beide Stoffwechselwege sind als monogene Ursachen für angeborene Lupuserkrankungen bekannt [4,5].
Primäre Immundefekte, die sich auch durch Autoimmunität manifestieren können, beinhalten meist Störungen der B-/T-Zell-Entwicklung und -Funktion, Komplementdefekte oder eine überschießende Inflammation durch Apoptosedefekte. Darüber hinaus existieren seltenere monogene Formen primärer Immundefizienz, welche zum Teil nur aus einzelnen Fallberichten bekannt sind und hier nicht vollständig diskutiert werden können.
Störungen der T-Zell-Entwicklung
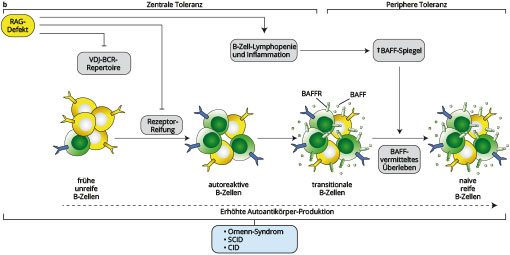
Die Rekombinasen RAG1 und RAG2 werden in der Entwicklung von Lymphozyten exprimiert. Im Rahmen der somatischen Rekombination vermitteln sie Doppelstrangbrüche in der DNA, wodurch aus dem Arsenal der variablen Genabschnitte V, D und J die Sequenz für die Antigen-bindenden Bereiche des Immunglobulin- sowie T-Zellrezeptors zusammengesetzt wird. Fehlt eines der Enzyme, führt dies zu einem schweren kombinierten Immundefekt (severe combined immunodeficiency, SCID) mit vollständig abwesenden B- und T-Zellen bei erhaltenen NK-Zellen. Klinisch fällt dies durch lebensbedrohliche Infektionen auf. Hypomorphe Varianten mit erhaltener Restaktivität der Enzyme bieten ein breites Spektrum klinischer Manifestationen. Ein Beispiel hierfür ist das Omenn Syndrom, welches durch Erythrodermie, Lymphadenopathie und Hepatosplenomegalie, sowie eine Infektneigung bei Hypogammaglobulinämie mit erhöhten IgE Spiegeln auffällt. Die wenigen generierten T-Lymphozyten sind kaum einer Kompetition um Überlebensfaktoren ausgesetzt, wodurch auch weniger affine autoreaktive T-Lymphozyten proliferieren können. Darüber hinaus sind auch regulatorische T-Zellen in Zahl und Funktion vermindert. Die gemeinsame Endstrecke sind Organinfiltrationen oligoklonaler, aktivierter T-Zellen ähnlich einer Graft-versus-Host-Reaktion.
Eine Störung der B-Zell-Reifung resultiert beim Omenn-Syndrom in einer B-Lymphopenie. Durch vermehrte Verfügbarkeit des B-cell activating factors (BAFF) kommt es zu einer gestörten negativen Selektion und Expansion von autoreaktiven B-Zellen [8]. Die Folge sind zirkulierende Autoantikörper, welche mit weiteren autoimmunen Manifestationen wie Alopezie, Myasthenia gravis, Vitiligo und Psoriasis einhergehen.
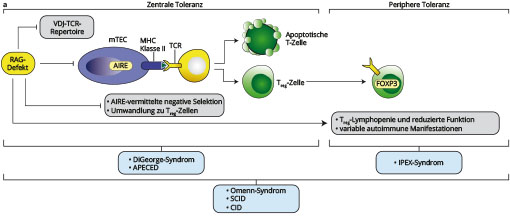
Auch Störungen der T-Zell-Reifung und der zentralen Toleranzentwicklung führen als gemeinsame Endstrecke zu Autoimmunität. Beispiele hierfür sind Mutationen im Gen für den Transkriptionsfaktor Autoimmun-Regulator (AIRE), welches die Autoantigenpräsentation im Thymus reguliert; im DNA-Reparaturenzym DCLRE1C/Artemis, welches im Rahmen der V(D)J-Rekombination die Reparatur des Doppelstrangbruches vermittelt; sowie das Mikrodeletionssyndrom 22q11, auch DiGeorge-Syndrom genannt, das zu einem hypoplastischen oder vollständig fehlenden Thymus mit konsekutiver Störung der T-Zellentwicklung führt.
Unzureichende T-Zell-Regulation
Neben der negativen Selektion autoreaktiver T-Zellen im Rahmen ihrer Entwicklung im Thymus ist auch die dämpfende Funktion regulatorischer T-Zellen (Tregs) notwendig, um eine überschießende Reaktion des Immunsystems gegen körpereigene Strukturen zu verhindern. Dies wird deutlich durch monogene Defekte, welche zu fehlenden oder nicht korrekt funktionierenden Tregs führen. Mutationen im auf dem X-Chromosom lokalisierten Gen für den Transkriptionsfaktor FOXP3 verursachen ein vorwiegend Männer betreffendes Polyautoimmunsyndrom, zu dessen häufigsten Manifestationen chronisch-entzündliche Darmerkrankung, Typ-1-Diabetes und Dermatitis gehören (immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome). Durch die in Anzahl und Funktion eingeschränkten regulatorischen T-Zellen kommt es zu einer Enthemmung der Immunantwort mit einer vermehrten Aktivierung und Proliferation insbesondere von TH2-Zellen und B-Zellen mit konsekutiver Produktion von Autoantikörpern.