
Der „Immunstatus“ auf dem Prüfstand
Stufendiagnostik von Immundefekten
Die moderne Diagnostik von Immundefekten umfasst neben Klinik und Basislabor spezielle durchflusszytometrische und genetische Untersuchungen inkl. Panel-Diagnostik mittels Next Generation Sequencing. Neben den häufigeren erworbenen Störungen sind inzwischen mehr als 300 angeborene Immundefekte bekannt, deren Komplexität nicht zuletzt darin besteht, dass einerseits unterschiedliche Gendefekte ähnliche klinische Phänotypen hervorrufen und andererseits verschiedene Phänotypen auf demselben Gendefekt beruhen können.
Schlüsselwörter: Immunstatus, Immundefekt, Durchflusszytometrie, Next Generation Sequencing
Der menschliche Organismus ist mit einem ausgefeilten Verteidigungsmechanismus ausgestattet, dessen Hauptfunktion darin besteht, fremde von körpereigenen Antigenen zu unterscheiden und eine entsprechende Immunantwort zu initiieren. Historisch gesehen wird die angeborene von der adaptiven Immunität unterschieden, wobei diese Systeme eng interagieren. Die Immunantwort wird sowohl von zellulären Komponenten (B- und T-Lymphozyten, NK-Zellen, Makrophagen/Monozyten, Granulozyten und antigen-präsentierende Zellen) als auch von humoralen Bestandteilen (Antikörper, Komplementfaktoren, Zytokine u. a. lösliche Effektormoleküle) vermittelt. Während die angeborene Immunantwort relativ unspezifisch, dafür schnell konservierte Strukturen von Pathogenen erkennt, reagiert das adaptive Immunsystem – vermittelt durch die extrem variablen B- und T-Zellrezeptoren – zeitverzögert, jedoch hochspezifisch. Zudem kann es ein immunologisches Gedächtnis ausbilden, welches bei erneutem Kontakt effizienter reagiert.
Es erstaunt nicht, dass Entwicklung und Funktion dieses hochkomplexen Systems an den unterschiedlichsten Stellen defekt sein und so zu einer erhöhten Infektanfälligkeit führen können. Grundsätzlich muss man bei allen Zuständen mit pathologischer Infektanfälligkeit primäre (angeborene) und sekundäre (erworbene) Immundefekte in Erwägung ziehen und differenzieren. Dieser Beitrag befasst sich vor allem mit der ersten Gruppe; zur letzteren gehören die erworbene Immunschwächekrankheit AIDS bei HIV-Infektionen, andere virale oder auch toxische Schädigungen des Immunsystems, Leukämien und Lymphome, Diabetes mellitus, zystische Fibrose u. v. m.
Angeborene Immundefekte
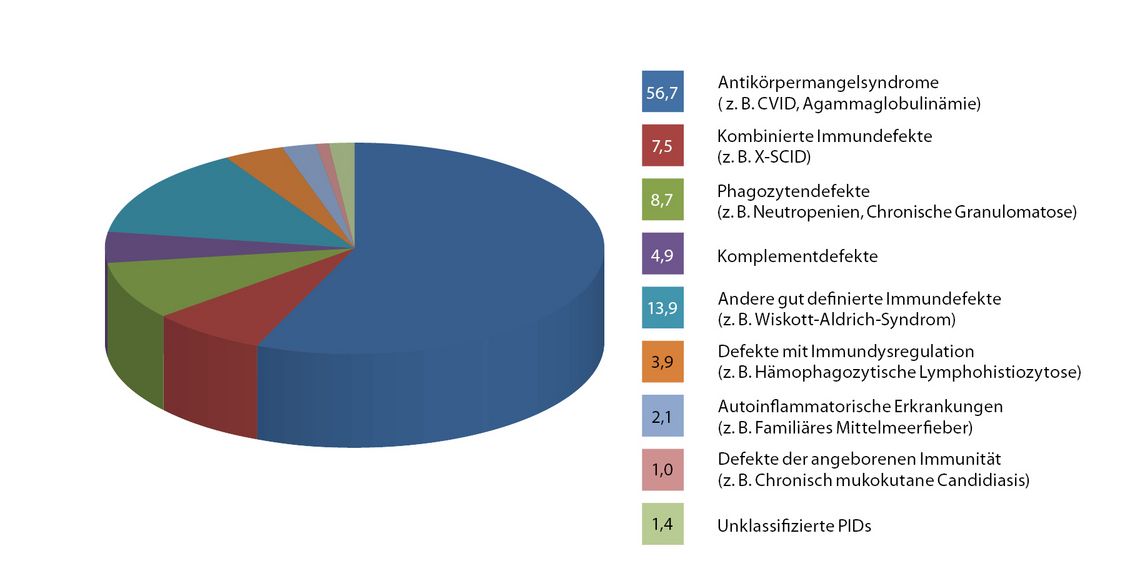
Über die Hälfte der Patienten mit einem primären Immundefekt (PID) leidet an Antikörpermangelzuständen, aber auch die zelluläre Abwehr durch T-Zellen oder Phagozyten kann betroffen sein; oftmals kommt es zu einer kombinierten Beeinträchtigung des zellulären und humoralen Systems (Abb. 1). Das Expertenkomitee der International Union of Immunological Societies aktualisiert in regelmäßigen Abständen einen umfassenden Katalog aller bekannten PIDs, die nach klinischen, immunologischen und genetischen Aspekten klassifiziert werden[1].
Die meisten angeborenen Formen manifestieren sich bereits im Kindesalter, werden aber bei einem nicht unerheblichen Teil der Patienten erst im Erwachsenenalter diagnostiziert. Besonders Antikörpermangelzustände wie CVID (common variable immunodeficiency) können erstmals in der zweiten oder dritten Lebensdekade evident werden. Einige Immundefekte treten zudem erst bei einer Infektion mit spezifischen Erregern, beispielsweise EBV bei X-chromosomalem lymphoproliferativem Syndrom, in Erscheinung.
Jeder einzelne der über 300 bekannten PIDs zählt zu den seltenen Erkrankungen, wobei heutige Schätzungen – basierend auf Daten aus den USA – von einer Gesamtprävalenz aller PIDs von etwa 1 : 2.000 ausgehen. Nach Angaben der Deutschen Selbsthilfe Angeborene Immundefekte e. V. (dsai) befinden sich in Deutschland derzeit aber nur ca. 3.500 PID-Patienten in Behandlung; die Dunkelziffer ist demnach hoch.
Klinische Kriterien
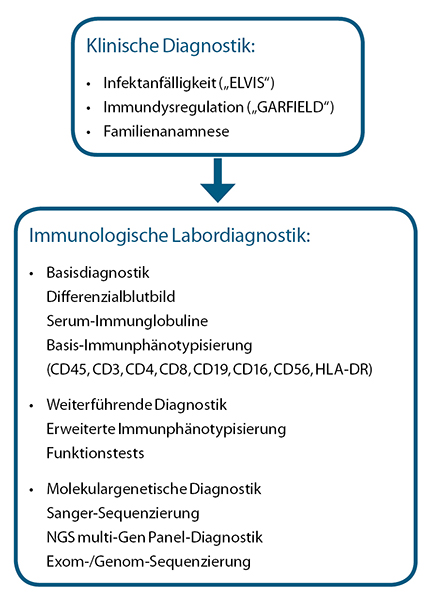
Die Diagnostik umfasst die in Abbildung 2 dargestellten Stufen[3]. Das klinische Hauptmerkmal ist eine erhöhte Infektanfälligkeit, die durch eine sorgfältige Eigen- und Familienanamnese sowie eine körperliche Untersuchung eruiert wird. Dies ist keineswegs trivial, denn die Infekthäufigkeit wird durch zahlreiche Faktoren wie soziale Strukturen, Familiengröße oder Besuch einer Kindertagesstätte beeinflusst, sodass es schwer möglich ist, einen oberen Grenzwert für die physiologische Infekthäufigkeit anzugeben[4]. Zusätzlich können primäre Immundefekte nicht nur mit wiederkehrenden, persistierenden und/oder schwer behandelbaren Infektionen, sondern auch mit Autoimmunität, Lymphoproliferation und malignen Erkrankungen assoziiert sein.
2011 erarbeitete die Arbeitsgemeinschaft Pädiatrische Immunologie (API) e. V. gemeinsam mit der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF) e. V. die Leitlinie „Diagnostik von primären Immundefekten“ und fasste darin Warnzeichen in Form der Akronyme ELVIS und GARFIELD zusammen. ELVIS bezieht sich dabei auf Zeichen einer pathologischen Infektanfälligkeit: Art des Erregers (atypische wie Pneumocystis jirovecii oder schwere Infektionen mit gewöhnlichen Erregern wie Pneumokokken), Lokalisation, Schwere der Infektion (Verlauf und Intensität) und Summe der Infektionen. GARFIELD umschreibt Zeichen einer Immundysregulation: Granulome, Autoimmunität, rezidivierendes Fieber, ungewöhnliche Ekzeme, Lymphoproliferation und chronische Darmentzündung.
Basislabor
Die Basisdiagnostik beinhaltet ein Differenzialblutbild, die Bestimmung der Serum-Immunglobuline (IgG, IgM, IgA und IgE) und eine Lymphozyten-Basisimmunphänotypisierung (Abb. 2). Lymphozytopenien oder Neutropenien sowie Thrombozytopenien können dabei bereits erste Hinweise für einen Immundefekt liefern.
Niedrige Zellzahlen gehen entweder auf eine verminderte Zellproduktion – zum Beispiel Entwicklungsarrest bei schweren kombinierten Immundefekten (SCID) oder Wiskott-Aldrich-Syndrom – oder auf ein verkürztes Überleben der Zellen (etwa bei familiärer Hämophagozytose) zurück. PIDs können auch mit erhöhten Zellzahlen wie Eosinophilien einhergehen, zum Beispiel bei Hyper-IgE-Syndrom, Omenn-Syndrom oder IPEX-Syndrom[4]. Neben quantitativen Blutbildveränderungen können qualitative morphologische Veränderungen auf einen primären Immundefekt hinweisen (zum Beispiel Howell-Jolly-Körperchen bei kongenitaler Asplenie, Mikrothrombozyten bei Wiskott-Aldrich-Syndrom oder Riesen-Granula bei Chediak-Higashi-Syndrom)[4, 5].
Da mehr als die Hälfte der Patienten mit primärem Immundefekt an einem Antikörpermangel leidet (Abb. 1), ist die Bestimmung der Immunglobuline im Serum ein wichtiger differenzialdiagnostischer Baustein. Nicht nur verminderte, sondern auch erhöhte Werte können auf einen Immundefekt hindeuten, insbesondere erhöhtes IgE (zum Beispiel bei Hyper-IgE-Syndrom, Omenn-Syndrom, IPEX-Syndrom), IgM (bei Immunglobulin-Klassenwechsel-Rekombinations-Defekten) oder IgG (bei Autoimmun-Lymphoproliferativem-Syndrom)[5]. Zur Beurteilung der Fähigkeit, spezifische Antikörper gegen Protein- und Polysaccharid-Antigene zu bilden, dient die Bestimmung von Impfantikörpern gegen Tetanustoxoid, Diphtherietoxoid und Pneumokokken-Polysaccharid (ggf. nach Boosterung). Die Untersuchung der IgG-Subklassen und des Komplementsystems bleibt speziellen Fragestellungen vorbehalten und muss nicht Bestandteil der Basisdiagnostik sein[5, 6].
Neben Blutbild und serologischer Diagnostik kommt der Durchflusszytometrie eine Schlüsselrolle in der Abklärung von Immundefekten zu (Abb. 3). Durch die kontinuierliche Weiterentwicklung dieser Technik können inzwischen immer kleinere und differenziertere Subpopulationen von Zellen charakterisiert und beurteilt werden. Im Allgemeinen ist ein durchflusszytometrisches 8-Farben-Basispanel einschließlich der Zelloberflächen-Marker CD45 (Panleukozyten-Marker), CD3 (T-Lymphozyten), CD4 (T-Helfer-Lymphozyten), CD8 (zytotoxische T-Lymphozyten), CD19 (B-Lymphozyten), CD16/CD56 (natürliche Killerzellen) und HLA-DR (Aktivierungsmarker) ausreichend, um die lymphatischen Hauptpopulationen zu differenzieren und quantifizieren[3].

Dr. rer. nat. Mirzokhid Rakhmanov (links)
Dr. rer. nat. Barbara Bangol (rechts)
Dr. med. Leon Holzscheiter (Mitte)
mirzokhid.rakhmanov@
medizinische-genetik.de
Zentrum für Humangenetik und
Laboratoriumsdiagnostik (MVZ)
Dr. Klein, Dr. Rost und Kollegen, München