Zwischen Gut und Böse
Zufallsbefund B-Zell-Lymphozytose
Auf der Skala der Interpretationsmöglichkeiten für eine erhöhte Lymphozytenzahl nimmt die MBL eine Mittelstellung zwischen Normvariante und maligner Neoplasie ein.
Sortiert man die täglichen Laborbefunde nach der Häufigkeit, so rangieren Blutbilder meist auf Platz 1. Und da heute nahezu alle Hämatologieautomaten eine Leukozytendifferenzierung anbieten (S. 164 ff.), stellen Lymphozytosen mit Absolutwerten über 3 G/l einen häufigen Zufallsbefund dar. Von einer relativen Lymphozytose spricht man, wenn der Lymphozytenanteil bei normaler Gesamtleukozytenzahl über 45% liegt. Das Spektrum der Interpretationsmöglichkeiten reicht von konstitutionellen Varianten ohne Krankheitswert über reaktive Erhöhungen – zum Beispiel bei Virusinfektionen – bis zu Non-Hodgkin-Lymphomen oder aggressiven akuten lymphatischen Leukämien.
Bei klinisch unklaren oder mit kritischen Warnmeldungen (Flags) versehenen Befunden der maschinellen Blutbildanalyse ist eine mikroskopische Zelldifferenzierung erforderlich. Häufig ergeben sich bereits aus der Morphologie Hinweise auf die Genese; so sieht man im Bild oben Lymphozyten mit grobscholliger, „fußballartiger“ Kernstruktur und zwei Kernschatten als (unspezifische) Hinweise auf eine mögliche Neoplasie. Die weitergehende Charakterisierung erfolgt dann mithilfe der durchflusszytometrischen Immunphänotypisierung aus Blut und Knochenmark. Gegebenenfalls schließen sich molekulardiagnostische Analysen bzw. eine Lymphknotenhistologie an.
Persistierende Lymphozytosen
Anhaltend erhöhte Lymphozytenzahlen sollten immer Anlass für eine umfassende Ursachenforschung sein, ehe man eine harmlose Normvariante annimmt. Dabei spricht eine Vermehrung von T- und NK-Zellen (Helfer-, Suppressor- und natürliche Killerzellen) eher für eine reaktive Lymphozytose, die auf eine ständige Immunaktivierung bei chronischen Erkrankungen einschließlich Autoimmunsyndromen und Tumoren hinweisen könnte.
Wesentlich seltener findet man eine persistierende polyklonale B-Zell-Lymphozytose (PPBL), die eine Mittelstellung zwischen konstitutionellen und potenziell präneoplastischen Veränderungen einnimmt. Diese spezielle Form der Lymphozytose tritt fast nur bei Frauen auf und imponiert im Blutausstrich recht auffällig durch zweikernige, zytoplasmareiche Lymphozyten. Sie ist häufig mit strukturellen oder numerischen Veränderungen des Chromosoms 3 vergesellschaftet und gilt als prädisponierend für lymphoproliferative Erkrankungen.
Monoklonale B-Lymphozytose
Im Gegensatz zu dieser seltenen polyklonalen Form kommt die monoklonale B-Lymphozytose (MBL) in bis zu 5% der Allgemeinbevölkerung vor[1]. Die Angaben zur Prävalenz schwanken methodenabhängig erheblich, da die MBL als „fakultative Präneoplasie“ fließende Übergänge zur chronisch lymphatischen Leukämie (CLL)[2] und anderen lymphoproliferativen Erkrankungen (z. B. Mantelzell-Lymphom, follikuläres Lymphom) aufweist. Die Prävalenz der MBL steigt mit zunehmendem Alter an und ist in Familien mit manifesten lymphoproliferativen Erkrankungen deutlich erhöht. Per se besitzt die MBL keinen Krankheitswert.
Die Diagnose basiert auf dem Nachweis einer monoklonalen B-Zell-Population, die über mindestens drei Monate persistiert und eines der folgenden Kriterien erfüllt:
• klonale Überexpression eines Leichtkettentyps (vorwiegend Kappa, sogenannte Leichtkettenrestriktion) oder
• monoklonales Schwerketten-Gen-Rearrangement oder
• niedrige bis fehlende Expression von Oberflächen-Immunglobulinen in mindestens 25% der Zellen oder
• aberranter Immunphänotyp (s. u.).
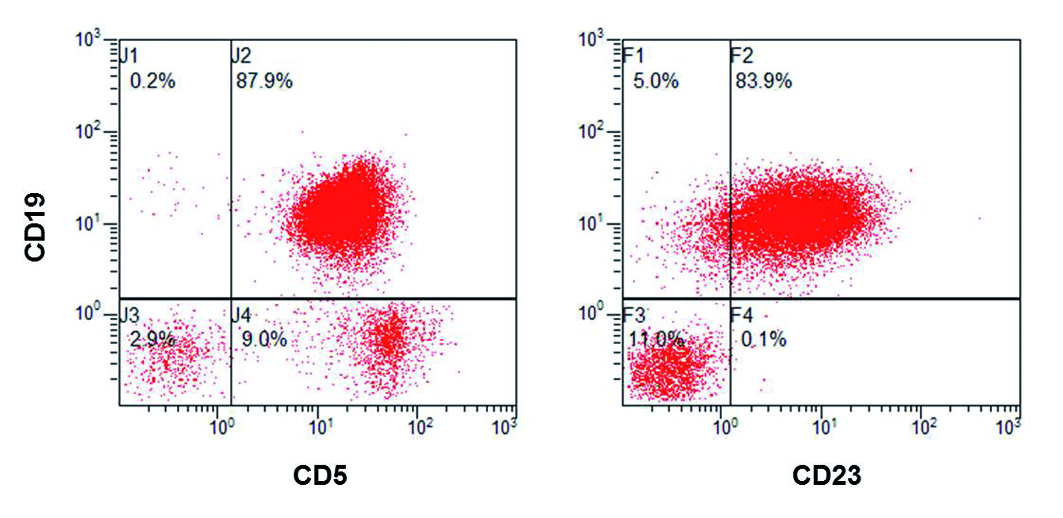
Für diese Diagnostik müssen verschiedenste methodische Register gezogen werden, insbesondere Durchflusszytometrie, Proteinchemie (Immunoassays oder Immunfixation für freie leichte Ketten) und evtl. Molekulardiagnostik (VDJ-Rekombinationsmuster, Zytogenetik). Im Durchflusszytometer weisen 80% aller Fälle einen CLL-ähnlichen Phänotypen (CD5+/CD19+/CD20low/CD23+/Iglow) auf; als Abweichungen sind v. a. CD5- (Non-CLL), CD20+, CD23- und Ig+ zu nennen. Auszuschließen ist eine MBL bei hoher Gesamtzahl der B-Lymphozyten (> 5 G/l) oder wenn bestimmte Differenzialdiagnosen (Autoimmunkrankheit, Infekt, Lymphadenopathie, manifeste lymphoproliferative Neoplasie) vorliegen.
Progressionsrisiko
1 bis 2% der MBL-Fälle gehen pro Jahr in eine behandlungsbedürftige CLL oder ein anderes malignes Lymphom über, und umgekehrt ist aufgrund der Ergebnisse großer epidemiologischer Studien anzunehmen, dass so gut wie jeder CLL eine MBL vorangeht. Eine Abschätzung der Progressionswahrscheinlichkeit ist kaum möglich, aber als ungünstig gilt das Ansteigen der monoklonalen B-Zell-Population über 1,9 G/l und die Zunahme typischer genetischer Läsionen wie etwa del(13q).
