Die akute lymphatische Leukämie im Erwachsenenalter – aktuelle Diagnostik und Therapie
Die akute lymphatische Leukämie (ALL) des Erwachsenenalters ist die zweithäufigste akute Leukämie bei Erwachsenen. Verschiedene chromosomale wie auch molekulargenetische Veränderungen führen zu einer Störung der Reifung und der Proliferation lymphatischer Vorläuferzellen. ALL-Erkrankungen des B-lymphozytären Typs überwiegen dabei deutlich gegenüber der ALL des T-lymphozytären Typs. Neben klinischen Parametern werden in jüngerer Zeit vermehrt genetische Faktoren zur Klassifikation und Risikoabschätzung bei der ALL verwendet. Das Rückgrat der Therapie bilden komplexe Chemotherapie-Protokolle; neben verschiedenen klassischen Zytostatika werden zunehmend auch immunologische Ansätze wie Antikörpertherapien und Stammzelltransplantationen, aber auch zielgerichtete Therapien eingesetzt. Ältere Patienten qualifizieren sich häufig nicht für intensive Therapieprotokolle und werden mit toxizitäts-adaptierten Therapieregimes behandelt.
Schlüsselwörter: Akute lymphatische Leukämie, Therapie, Diagnose
Die akute lymphatische Leukämie (ALL) ist durch eine maligne klonale Proliferation lymphozytärer Vorläuferzellen des B- oder T-Zelltyps gekennzeichnet. Die ALL kann sowohl im Kindesalter als auch im Erwachsenenalter auftreten und repräsentiert speziell im Erwachsenenalter eine komplexe Erkrankung, die mit multimodalen Ansätzen behandelt wird. Aktuelle Entwicklungen in der Diagnostik und der Therapie der ALL haben in jüngerer Zeit zu einer Erweiterung der Therapieoptionen und zu einer Verbesserung der Prognose der Patienten geführt. Im Folgenden werden aktuelle Daten und Therapiekonzepte zusammengefasst dargestellt.
Epidemiologie
Die ALL hat gemäß aktueller Daten der US-amerikanischen SEER-Datenbank eine Gesamtinzidenz von ca.
1,7 Neuerkrankungen/100.000 Einwohner [1]. Damit ist die ALL für 0,4% aller Krebsfälle in den USA verantwortlich. Ähnliche Zahlen liegen für Deutschland und Europa vor. Obwohl die Erkrankungsrate der ALL über die letzten
40 Jahre minimal anzusteigen scheint, nimmt die Rate an ALL-assoziierten Sterbefällen deutlich ab. Letzteres spiegelt die Verbesserung der Therapieoptionen wider. Betrachtet man die Therapieergebnisse über die Jahre 1975–2014 zusammenfassend, so ist festzuhalten, dass die 5-Jahre-Überlebensrate sich von 31% im Jahr 1975 über die Zeit auf 69% im Jahr 2009 verbessert hat [1]. In den USA ist ein Anteil an ALL-bedingten Todesfällen von 0,4/100.000 Einwohner pro Jahr zu verzeichnen [1].
Über 50% der ALL-Fälle werden im Lebensalter von unter 20 Jahren diagnostiziert. Die ALL ist somit eine häufige Neoplasie im Kindesalter. Im Erwachsenenalter kann sie sowohl in der Adoleszenz als auch bis hin ins hohe Lebensalter auftreten. Das mediane Alter bei Diagnosestellung liegt bei 15 Lebensjahren. Männer scheinen etwas häufiger an einer ALL zu erkranken als Frauen.
Pathogenese
Wesentliches Charakteristikum der Pathogenese der ALL ist die autonome Proliferation und abnorme Differenzierung einer klonalen Zellpopulation mit lymphatischem Phänotyp. Verschiedene genetische Syndrome, welche zu der Entwicklung einer ALL prädisponieren, konnten identifiziert werden [2]. Hierzu gehören beispielsweise das Down-Syndrom, die Fanconi-Anämie, das Bloom-Syndrom und auch die Ataxia teleangiectasia. Andere Risikofaktoren umfassen eine Infektion mit Epstein-Barr- oder dem humanen Immunodefizienz-Virus sowie die Exposition gegenüber Bestrahlung oder Pestiziden. In einem Großteil der Fälle können jedoch keine spezifischen prädisponierenden Faktoren identifiziert werden.
Es gibt eine Vielzahl chromosomaler Aberrationen, die bei der ALL auftreten können. Charakteristische Veränderungen umfassen u. a. die Translokation t(9;22), Rearrangements des MLL-Gens sowie die Translokation t(12;21) [3]. Neuere Untersuchungen haben gezeigt, dass u. a. genetische Veränderungen der
IKAROS-Transkriptionsfaktor-Familie das Auftreten einer ALL begünstigen [4]). Weitere genetische Veränderungen entwickeln sich oftmals im Verlauf der Erkrankung [5]. Neue diagnostische Methoden wie Hochdurchsatz-Genanalysen mittels Next Generation Sequencing (NGS) werden gegenwärtig intensiv angewandt, um die komplexen molekulargenetischen Veränderungen der ALL zu analysieren [6].
Klinisch manifestiert sich die ALL in der Regel mit Symptomen einer Knochenmarksinsuffizienz. Führende Beschwerden sind oftmals durch Neutropenien bedingte Infektionen, eine Anämie-Symptomatik (Müdigkeit, Dyspnoe) oder auch Blutungszeichen. Insgesamt kann das klinische Bild dabei häufig unspezifisch sein. B-Symptome (Fieber, Nachtschweiß, Gewichtsverlust) sind möglich. Extramedulläre Manifestationen können Lymphknotenschwellungen, Splenomegalie und Hepatomegalie sein. Eine ZNS-Beteiligung bei Diagnosestellung tritt bei 5–8% der Patienten auf und erfordert spezielle Therapiestrategien. Akute lymphatische Leukämien vom T-Zell-Typ können durch mediastinale Raumforderungen imponieren.
Diagnostik
Die Diagnose der ALL wird in der Regel durch eine Untersuchung des Knochenmarks und des peripheren Blutes gestellt. Aspekte, welche in die Klassifikation der ALL eingehen, sind die Zytologie, die Morphologie, die Immunphänotypisierung sowie die zyto- und molekulargenetischen Befunde. Hierauf basieren Diagnose und Risikoklassifikation. Die aktuelle WHO-Klassifikation für akute lymphatische Leukämien ist in Tab. 1 dargestellt [7].
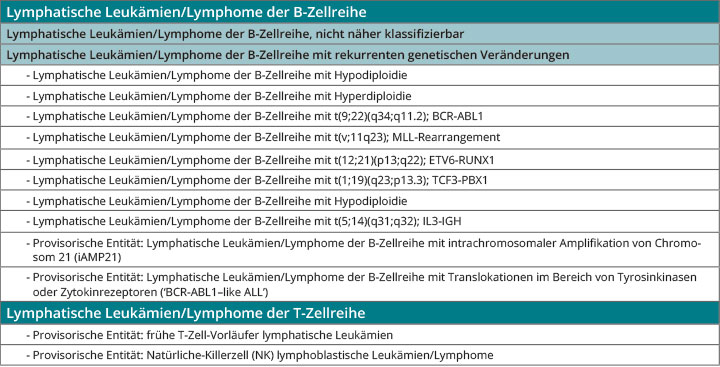
Die aktuelle WHO-Klassifikation behält die Gruppierung der ALL in B- und T-ALL bei [7]. Bei der B-ALL werden anhand ihrer genetischen Veränderungen weitere Subtypen unterschieden und noch nicht anderweitig spezifizierten ALL und Lymphomen der B-Zell-Reihe gegenübergestellt. Im Jahr 2016 wurden vier neue provisorische Entitäten hinzugefügt. Ebenso wurden die hypodiploide und die hyperdiploide ALL neu definiert. Die Französisch-Amerikanisch-Britische (FAB)-Klassifikation hat heutzutage keine Bedeutung mehr.
Eine Lumbalpunktion wird regelhaft initial durchgeführt, um einen ZNS-Befall auszuschließen. Sollte der Verdacht auf einen ZNS-Befall bestehen, ist zusätzlich die Durchführung von bildgebenden Verfahren wie der Magnetresonanztomografie indiziert. Weitere diagnostische Maßnahmen umfassen das Differenzialblutbild, die Bestimmung der Gerinnungsparameter und weiterführende Serumuntersuchungen. Ein Tumorlyse-Syndrom sollte stets ausgeschlossen werden (Harnsäure, Kreatinin, Kalzium, Phosphat, Gerinnung).
Prognose
Die Behandlung der ALL wird wesentlich durch eine adäquate Risikostratifikation geleitet. Dabei beeinflusst die Risikoeingruppierung sowohl die Auswahl des initialen Therapieregimes als auch die Hinzunahme spezieller Therapien wie der allogenen Stammzelltransplantation. Verschiedene Risikofaktoren wurden über die vergangenen Jahrzehnte identifiziert. Hierzu gehören u. a. das Alter des Patienten und die Leukozytenzahl zum Zeitpunkt der Erstdiagnose. Dabei zeigt sich, dass ein höheres Erkrankungsalter mit einer schlechten Prognose assoziiert ist. Besonders Patienten im Alter von über 60 Jahren haben eine eher schlechte Prognose mit einer Langzeit-Überlebensrate von nur 10–15% [8].
Wesentliche prognostische Bedeutung hat zunehmend die molekulare Diagnostik. Eine ALL mit der zytogenetischen Veränderung einer Translokation t(9;22) hatte vor der Einführung der Tyrosinkinase-Inhibitoren und der konsequenten Durchführung einer allogenen Stammzelltransplantation eine sehr schlechte Prognose [9]. Diese Therapieergebnisse konnten nunmehr deutlich verbessert werden [10, 11].
Ein weiterer wesentlicher prognostischer Faktor ist das Ansprechen auf die initiale Therapie. In verschiedenen Studien konnte sowohl bei der pädiatrischen als auch bei der ALL des Erwachsenen gezeigt werden, dass die Persistenz von Blasten im Knochenmark zu einem frühen Zeitpunkt der Therapie mit einer negativen Prognose einhergeht. Speziell die Deutsche ALL-Studiengruppe (GMALL) hat die Bedeutung der minimalen Resterkrankung (MRD) intensiv untersucht und ihren Stellenwert definiert [12]. Methoden, die hierbei zur Anwendung kommen, umfassen die Durchflusszytometrie und die Polymerase-Kettenreaktion. Brüggemann et al. konnten innerhalb der GMALL-Patienten mit Standardrisiko auf Basis des MRD-Niveaus Untergruppen mit niedrigem (0%), mittlerem (47%) und hohem Rezidivrisiko (94%) definieren [13]. Ähnliche Ergebnisse kommen aus der spanischen Studiengruppe [14].
Ungeklärt ist gegenwärtig die Frage nach der besten Therapie für die Altersgruppe der Adoleszenten und der jungen Erwachsenen (AYA; [15,16]). Diese Altersgruppe wird häufig vom 15. bis zum
39. Lebensjahr definiert. Verschiedene Daten legen nahe, dass pädiatrische Therapieprotokolle bei diesen Patienten eventuell bessere Therapieergebnisse erzielen [17]. Inwiefern dies tatsächlich der Fall ist, wird gegenwärtig geprüft. Es scheint, als ob die statistische Berücksichtigung der zugrunde liegenden genetischen Veränderungen die besseren Behandlungsergebnisse bei Verwendung pädiatrischer ALL-Protokolle nivellieren würde. AYA-Patienten, die in den Erwachsenen-Studien behandelt wurden, hatten oftmals andere genetische Veränderungen als die Patienten in den pädiatrischen Studien.
Therapieverfahren
Die Therapie der ALL ist komplex und umfasst klassische chemotherapeutische Ansätze mit verschiedenen Kombinationsregimes, immunologische Therapieansätze mit Antikörpern und zellulären Therapieverfahren sowie Elemente der Strahlentherapie.
Die Chemotherapie-Protokolle beinhalten in der Regel eine Induktionsphase, gefolgt von Konsolidierungs- sowie Reinintensivierungs-Zyklen, und eine Erhaltungstherapie. Die ZNS-Prophylaxe ist wesentlicher Bestandteil der ALL-Therapie [18]. Das Ziel der Induktionsphase ist die komplette hämatologische Remission mit dem möglichst frühzeitigen Erreichen einer MRD-Negativität. Das wesentliche Rückgrat der Therapie besteht aus Kortikosteroiden, Vinca-Alkaloiden und Anthrazyklinen. Weitere Substanzen, die in der Therapie der ALL verwendet werden, sind die L-Asparaginase, Cyclophosphamid und Cytarabinosid [2]. Die Hinzunahme von Tyrosinkinase-Inhibitoren bei der Philadelphia-Chromosom-positiven ALL ist inzwischen Standard. Im Bereich der T-ALL ist Nelarabin eine sehr aktive Substanz.
Therapieverfahren auf immunologischer Basis sind ein weiterer wesentlicher Bestandteil der ALL-Therapie. Während der Stellenwert der allogenen Stammzelltransplantation seit Jahren bekannt ist, hat die Einführung von Antikörper-basierten wie auch von zellulären Therapieverfahren in jüngster Zeit den Bereich immunologischer Optionen bei der
B-ALL deutlich erweitert. Die verschiedenen Ansätze sind in Abb. 1 schematisch dargestellt.
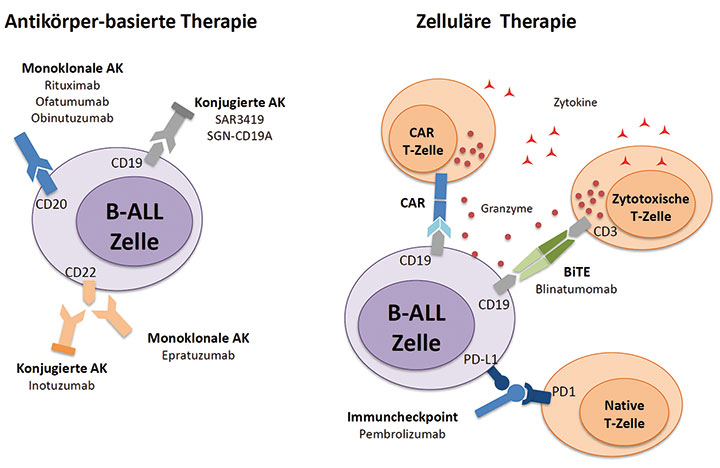
Antikörper-basierte Therapien zielen auf Zelloberflächen-Antigene wie CD20, CD52, CD19 und CD22 und bestehen aus den Antikörpern entweder in reiner Form oder in Kombination mit einem konjugierten Toxin. Diesen Therapien gegenüberzustellen sind zelluläre Therapiekonzepte, die mit bispezifischen Antikörpern (BiTE) oder mit genetisch modifizierten T-Zellen, die einen chimären Antigen-Rezeptor (CAR-T) tragen, durchgeführt werden [19].
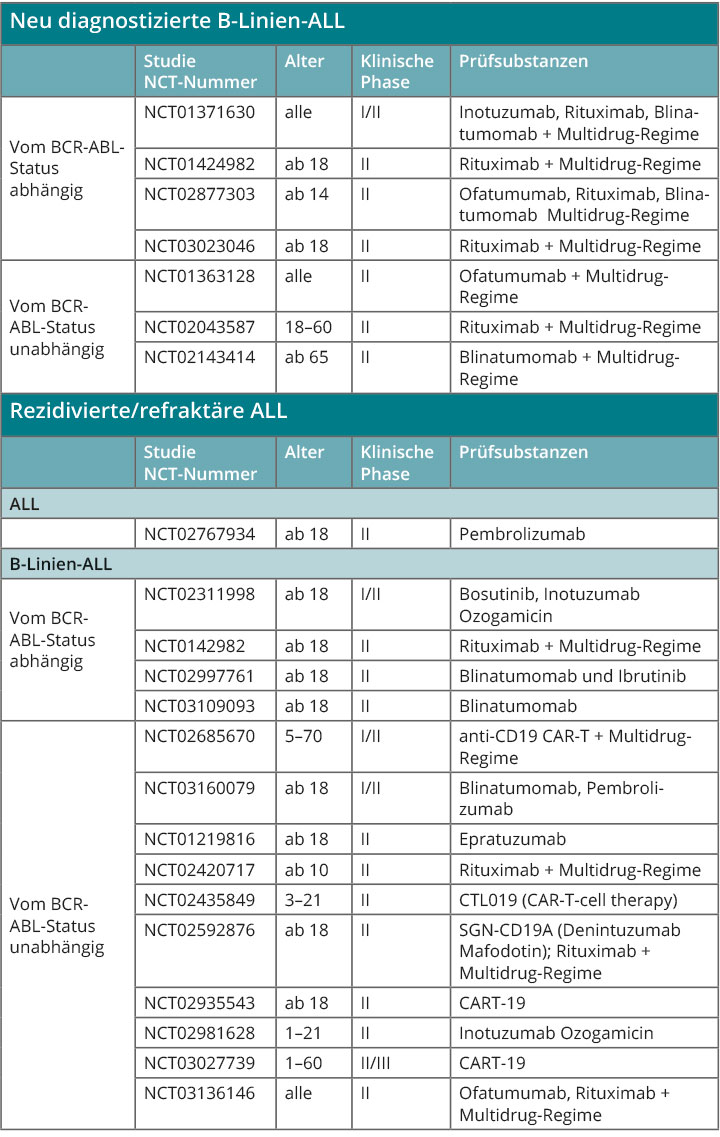
Spezifische Antikörpertherapien wie die Behandlung mit Rituximab bei der CD20-positiven ALL sind inzwischen etabliert [20]. Neue konjugierte und unkonjugierte Antikörpertherapien werden gegenwärtig in klinischen Studien untersucht. Zugelassen ist seit Kurzem zur Behandlung der rezidivierten CD22-positiven ALL das Immunkonjugat Inotuzumab Ozogamicin. Im Bereich der Therapieverfahren auf zellulärer Basis haben Studien eine wesentliche Effektivität des bispezifischen Antikörpers Blinatumomab gezeigt. Er ist sowohl gegen das Oberflächenantigen CD19 auf der B-ALL-Zelle als auch gegen den CD3-Rezeptor von T-Zellen gerichtet. T-Zellen mit chimärem Antigenrezeptor (CAR-T-Zellen) haben, basierend auf den sehr positiven Daten der frühen klinischen Studien, eine FDA-Zulassung für die pädiatrische ALL erhalten [21]. Die Verfügbarkeit dieser Therapieform in Deutschland ist gegenwärtig limitiert, wird aber in den kommenden Monaten zunehmen, da die entsprechende Logistik aktiv aufgebaut wird. Eine Auswahl aktueller Studienkonzepte für die neu diagnostizierte wie für die rezidivierte B-ALL ist in Tab. 2 dargestellt.
Rezidivierte oder refraktäre Erkrankungen
Patienten mit rezidivierter oder refraktärer ALL haben eine sehr schlechte Prognose. Diese Patienten sollten innovativen Therapieverfahren im Rahmen von Studien zugeführt werden (u. a. Studien der GMALL-Studiengruppe und Tab. 2; [18]). Die konventionelle Therapie umfasst die Applikation einer Chemoimmuntherapie, in der Regel gefolgt von einer allogenen Stammzelltransplantation. Letzteres Therapieverfahren ist dabei wesentlich – es stellt in dieser Situation den einzigen kurativen Therapieansatz dar. Jüngere Daten legen nahe, dass mit Substanzen wie Blinatumomab ebenfalls das rezidivfreie Überleben verlängert werden kann, wenn die Therapie bei niedrigem MRD-Niveau begonnen wird. Diese Daten unterstreichen, wie wichtig die regelmäßige Kontrolle der minimalen Resterkrankung ist.
Schlussfolgerung
Die ALL im Erwachsenenalter ist eine seltene Erkrankung, die eine komplexe Therapie erfordert. Hierbei ist eine umfassende Diagnostik, u. a. um das genetische Risikoprofil der Patienten zu erheben, bedeutsam. Die anschließende Therapie ist intensiv und mit signifikanter Toxizität assoziiert. Durch multimodale Therapieverfahren konnte die Langzeitprognose der ALL-Patienten allerdings deutlich verbessert werden, sodass heutzutage mehr als 50% der Patienten länger als fünf Jahre überleben. Insbesondere bei älteren sowie auch bei rezidivierten und refraktären Patienten ist die Auswahl der Therapie vom Zustand des Patienten als auch von den bisherigen dosislimitierenden Nebenwirkungen abhängig. Neue zielgerichtete Therapieverfahren scheinen hier vielversprechend. Aufgrund der großen genetischen Heterogenität der ALL scheinen immunologische Ansätze gegenwärtig oftmals breiter einsetzbar als zielgerichtete Therapien. Man darf gespannt sein, inwiefern diese innovativen Therapieverfahren in die neuen Therapiestrategien involviert werden können.
Literatur
1. Cancer Statistics Review, 1975-2014 - SEER Statistics. Available from: seer.cancer.gov/csr/1975_2014/ (cited 2017, Sep 6).
2. Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J 2017; 7: e577.
3. Mullighan CG et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007; 446: 758-64.
4. Kobitzsch B et al. Loss-of-function but not dominant-negative intragenic IKZF1 deletions are associated with an adverse prognosis in adult BCR-ABL-negative acute lymphoblastic leukemia. Haematologica 2017; Jul 27 [Prepub ahead of print, DOI 10.3324/haematol.2016.161273].
5. Mullighan CG et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008; 322: 1377-80.
6. Mullighan CG. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematology 2014; 2014: 174-80.
7. Arber DA et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127: 2391-405.
8. Rowe JM. Prognostic factors in adult acute lymphoblastic leukaemia. Br. J. Haematol 2010; 150: 389-405.
9. Fielding AK et al. Prospective outcome data on 267 unselected adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia confirms superiority of allogeneic transplantation over chemotherapy in the pre-imatinib era: results from the International ALL Trial MRC UKALLXII/ECOG2993. Blood 2009; 113: 4489-96.
10. Ottmann O et al. Dasatinib induces rapid hematologic and cytogenetic responses in adult patients with Philadelphia chromosome positive acute lymphoblastic leukemia with resistance or intolerance to imatinib: Interim results of a phase 2 study. Blood 2007; 110: 2309-15.
11. Ravandi F et al. Long-term follow-up of a phase 2 study of chemotherapy plus dasatinib for the initial treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer 2015; 121: 4158-64.
12. Bruggemann M et al. Has MRD monitoring superseded other prognostic factors in adult ALL? Blood 2012; 120: 4470-81.
13. Bruggemann M et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood 2005; 107: 1116-23.
14. Ribera JM et al. Treatment of high-risk Philadelphia chromosome-negative acute lymphoblastic leukemia in adolescents and adults according to early cytologic response and minimal residual disease after consolidation assessed by flow cytometry: Final results of the PETHEMA ALL-AR-03 trial. J Clin Oncol 2014; 32: 1595-604.
15. Rytting ME et al. Acute lymphoblastic leukemia in adolescents and young adults. Cancer 2017; 123: 2398-403.
16. Curran E, Stock W. How I treat acute lymphoblastic leukemia in older adolescents and young adults. Blood 2015; 125: 3702-10.
17. Aldoss IT et al. Treatment of acute lymphoblastic leukemia in adults: Applying lessons learned in children. Oncology 2016; 30: 1080-91.
18. Kompetenz bei akuten und chronischen Leukämien. Available from: www.kompetenznetz-leukaemie.de/content/home/ (cited 2017, Sep 9).
19. Wei G et al.. Novel immunotherapies for adult patients with B-lineage acute lymphoblastic leukemia. J Hematol Oncol 2017; 10: 150.
20. Maury S et al. Rituximab in B-lineage adult acute lymphoblastic leukemia. N Engl J Med 2016; 375: 1044-53.
21. FDA approval brings first gene therapy to the United States. Office of the Commissioner; available from: www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm574058.htm (cited 2017 Sep 6).
22. ClinicalTrials.gov [Internet]. [cited 2017 Sep 6]. Available from: clinicaltrials.gov

Dr. rer. nat. Catrin Roolf

Dr. med Christina Große-Thie

Prof. Dr. med. Christian Junghanß
Zentrum für Innere Medizin
Klinik III – Hämatologie, Onkologie und Palliativmedizin
Universitätsmedizin Rostock
Ernst-Heydemann-Str. 6, 18057 Rostock
+49 381 494 7420