Neues zu Sarkomen
ASCO 2016
Weichteilsarkome sind seltene mesenchymale Tumoren, von denen pro Jahr und pro 100.000 Personen ungefähr zwei bis drei Fälle diagnostiziert werden. Die mediane Überlebenszeit der Patienten mit fortgeschrittenen bzw. metastasierten Weichteilsarkomen liegt meist unter einem Jahr, aber in den letzten Jahren wurden immerhin eine Reihe neuer Substanzen in die Klinik eingeführt. Beim diesjährigen ASCO-Kongress gab es einige interessante Abstracts, in denen durchaus relevante Weiterentwicklungen der Therapie mit signifikanter Verbesserung der Prognose präsentiert wurden.
Anlotinib hoch wirksam beim alveolären Weichteilsarkom
Anlotinib ist ein Multikinase-Inhibitor, der unter anderen die Rezeptoren für vaskulären endothelialen Wachstumsfaktor (VEGFR1/2/3), Fibroblasten-Wachstumsfaktor (FGFR1/2/3), Platelet-Derived Growth Factor (PDGFRα/β), c-Kit und Ret hemmt. Da es in China für Patienten mit Weichteilsarkomen, bei denen eine initiale Chemotherapie versagt hat, keine weitere zugelassene Standardtherapie gibt, wurde Anlotinib dort in einer Phase-II-Studie bei insgesamt 166 Patienten mit Weichteilsarkomen verschiedenster Histologien getestet, darunter Synovialsarkome (28,3%), Leiomyosarkomen (15,7%) und maligne fibröse Histiozytome (11,5%; [1]). Die Dosierung, die in einer vorhergehenden Phase-I-Studie gefunden worden war, betrug 12 mg/d und wurde in dreiwöchigen Zyklen gegeben – zwei Wochen lang Therapie, gefolgt von einer Woche Pause.
Primärer Endpunkt war das progressionsfreie Überleben (PFS) nach zwölf Wochen, wobei 57,2% aller Patienten dieses Ziel erreichten; die Krankheitskontrollrate zu diesem Zeitpunkt lag bei 73,5%, obwohl die Ansprechrate mit 11,5% relativ niedrig gewesen war. Die mediane progressionsfreie Überlebenszeit betrug 5,6 Monate. Patienten mit bestimmten Subtypen von Sarkomen profitierten besonders stark von der Therapie, so Yihebali Chi, Beijing: In der Gruppe von Patienten mit Leiomyosarkomen waren nach zwölf Wochen noch 69,1% progressionsfrei am Leben (Medianwert 11,1 Monate), in der Subgruppe mit alveolaren Weichteilsarkomen sogar 76,9% (Medianwert noch nicht erreicht), und auch bei einer Reihe anderer Entitäten lag die 12-Wochen-PFS-Rate oberhalb von 60%. Die alveolären Tumoren hatten mit 46,2% auch die höchste Ansprechrate.
An Grad-3/4-Nebenwirkungen traten vor allem ein Hypertonus, eine Hypertriglyzeridämie und ein Pneumothorax auf, aber die Raten waren mit 5–8% niedrig. Häufigere Toxizitäten vom Grad 1 und 2 waren außerdem Hand-Fuß-Syndrome, Proteinurie und Diarrhöen. 24 Patienten (14,5%) mussten wegen Nebenwirkungen die Dosis reduzieren.
Diese Ergebnisse sind vielversprechend; insbesondere scheinen Patienten mit alveolären Weichteilsarkomen von Anlotinib zu profitieren. Um das abzusichern, ist in China derzeit eine Phase-III-Studie im Gang, die 318 Patienten mit Chemotherapie-refraktären Weichteilsarkomen einschließen und in der Anlotinib randomisiert gegen Placebo geprüft werden soll [2]. Daneben wird die Substanz bei einer ganzen Reihe weiterer Tumorentitäten geprüft.
Lebensqualität auch bei Progression unter Eribulin höher
Das moderne Spindelgift Eribulin ist zur Behandlung des fortgeschrittenen, zuvor chemotherapierten Liposarkoms zugelassen. Die Zulassung basiert auf einer Phase-III-Studie, in der Eribulin bei 452 Patienten mit Lipo- oder Leiomyosarkomen das Gesamtüberleben gegenüber einer Dacarbazin-Monotherapie um zwei Monate verlängern konnte (von median 11,5 auf 13,5 Monate; Hazard Ratio 0,77; p = 0,0169; [3]). Grad-3/4-Nebenwirkungen waren im Eribulin-Arm etwas häufiger gewesen als im Dacarbazin-Arm (67% vs. 56%) – deshalb sollte in einer exploratorischen Analyse, die Stacie Hudgens,
Tucson, beim ASCO-Kongress vorstellte, untersucht werden, wie sich die Lebensqualität der Patienten bei Progression unter der jeweiligen Therapie entwickelt hatte [4].
Bei insgesamt 400 Patienten (209 im Eribulin-, 191 im Dacarbazin-Arm) kam der EORTC QLQ-C30-Fragebogen zum Einsatz. Zu Beginn der Studie unterschieden sich beide Arme nicht, zum Zeitpunkt der Progression war der globale Gesundheitszustand im Eribulin-Arm mit 62,1 Punkten signifikant besser als im Kontrollarm (56,1 Punkte; p = 0,0083). Auch bei körperlicher Funktion (73,3 vs. 63,9 Punkte; p = 0,0022), Nausea/Erbrechen (p = 0,0009) und Appetitverlust (p = 0,001) war der Mikrotubuli-Hemmstoff signifikant überlegen. Die Ergebnisse ähnelten sich in den beiden histologischen Untergruppen (Lipo- und Leiomyosarkome).
Eribulin bringt für diese Patienten also nicht nur die Chance einer signifikanten Lebensverlängerung gegenüber einer konventionellen Chemotherapie, sondern ist zugleich mit einer signifikant geringeren Einschränkung der Lebensqualität assoziiert, so Hudgens.
Pazopanib und Gemcitabin in Kombination besonders wirksam
Der Angiogenese-Inhibitor Pazopanib ist aufgrund der positiven Daten zum progressionsfreien Überleben in der placebokontrollierten Phase-III-Studie PALETTE zur Behandlung von Patienten mit nicht-adipozytischen Weichteilsarkomen nach Versagen der Chemotherapie zugelassen [5]. Weil auch Gemcitabin in der Monotherapie ebenso wie in Kombination mit Pazopanib Wirkung gezeigt hat [6], wurde diese Kombination in einer randomisierten deutschen Phase-II-Studie mit einer Pazopanib-Monotherapie verglichen [7].
Primärer Endpunkt war das progressionsfreie Überleben nach drei Monaten, und hier zeigte sich, wie Hans-Joachim Schmoll, Halle, in Chicago berichtete, bei den 86 auswertbaren Patienten ein signifikanter Vorteil für die Kombination mit 73,2% versus 45,5% (HR 1,62; p = 0,005). Die mediane Dauer des progressionsfreien Überlebens war mit 5,6 versus 1,9 Monaten ebenfalls signifikant verlängert
(HR 0,54; p = 0,01), in der Subgruppe mit Liposarkomen fiel der Unterschied mit 8,6 versus 1,5 Monaten noch drastischer aus, war aber – vermutlich aufgrund der kleinen Patientenzahlen – nicht mehr signifikant. Auch beim Gesamtüberleben zeigte sich bei den Liposarkomen ein numerischer Vorteil für die Kombination (median 25,4 vs. 11,1 Monate), im Gesamtkollektiv war kein signifikanter Unterschied zu erkennen, sehr wahrscheinlich bedingt durch die häufige Zugabe von Gemcitabin im Monotherapie-Arm, sobald die Patienten progredient wurden.
Diese Ergebnisse, so Schmoll, sind – nicht nur, aber vor allem für die Patienten mit Liposarkom – sehr vielversprechend und stellen eine Rationale dafür dar, diese Kombination trotz höherer Toxizität auch in der Erstlinientherapie von Weichteilsarkomen zu untersuchen.
Langes Überleben mit Regorafenib bei Leiomyosarkomen und Synovialsarkomen
Regorafenib ist ein Multikinase-Inhibitor, der neben BRAF auch verschiedene Angiogenese-Signalwege hemmt und bei Weichteilsarkomen mit Ausnahme von Liposarkomen das progressionsfreie Überleben verlängert. In der randomisierten Phase-II-Studie REGOSARC wurde Regorafenib bei Patienten mit Anthrazyklin-vorbehandelten metastasierten Weichteilsarkomen gegen Placebo getestet. Wie Nicolas Penel, Lille, in Chicago berichtete, hatten fast alle der 181 Patienten bereits Doxorubicin erhalten, 107 waren auch mit Ifosfamid, 70 mit Trabectedin und sechs mit Pazopanib vorbehandelt [8].
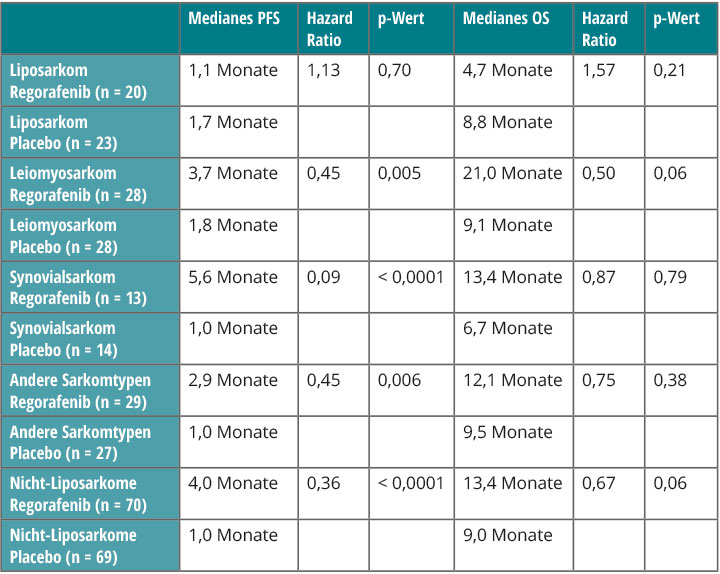
Mit vier partiellen Remissionen war die Ansprechrate niedrig, dafür fielen aber – abgesehen von den Liposarkomen – progressionsfreies und Gesamtüberleben sehr vielversprechend aus (Tab. 1). Vor allem beim Leiomyosarkom, aber auch beim Synovialsarkom waren die Überlebenszeiten erstaunlich hoch; dass das bei letzterer Subgruppe nicht signifikant war, ist vermutlich den niedrigen Patientenzahlen geschuldet. Die häufigsten Toxizitäten unter Regorafenib waren Asthenie, Anorexie, Diarrhö, Mukositis, arterieller Hochdruck und ein Hand-Fuß-Syndrom.
Ewing-Sarkom: Rolle für die Hochdosistherapie
Beim Ewing-Sarkom mit Lungen- oder Pleurametastasen wird nach einer Induktionschemotherapie üblicherweise eine Konsolidierung mit acht Zyklen Vincristin, Actinomycin D und Ifosfamid und einer Strahlentherapie für die pulmonale Manifestation gegeben. In einer europaweiten Phase-III-Studie unter Beteiligung von vier großen Studiengruppen wurde untersucht, ob man durch eine Hochdosistherapie mit Stammzellsupport bei Patienten, die schlecht auf eine Chemotherapie ansprechen oder sehr große Tumoren haben, die Lungenbestrahlung vermeiden kann, ohne an onkologischer Wirksamkeit einzubüßen. In dem sehr langen Zeitraum zwischen 2000 und 2014 wurden insgesamt 477 Patienten mit
Ewing-Sarkom und Lungen- oder Pleura-, aber keinen sonstigen Fernmetastasen als Hochrisiko-Patienten eingestuft, aber nur 216 von ihnen konnten randomisiert werden. Sie erhielten zunächst sechs Zyklen einer Induktionstherapie mit VIDE (Vincristin, Ifosfamid, Doxorubicin, Etoposid) und einen Zyklus VAI (Vincristin, Actinomycin D, Ifosfamid). Dann wurden sie randomisiert: Entweder erhielten sie in konventioneller Weise sieben weitere Zyklen VAI und eine Lungenbestrahlung oder eine Hochdosistherapie mit Busulfan und Melphalan und autologer Stammzelltransplantation [9].
Nach median acht Jahren Nachbeobachtung errechnete sich das ereignisfreie Überleben nach drei Jahren zu 66,9% in der Hochdosistherapie- und zu 53,1% in der Kontrollgruppe, so Jeremy Whelan, London (HR 0,63; p = 0,023). Auch beim Gesamtüberleben war die strahlenfreie Therapie erfolgreicher mit 77,8% versus 69,9% nach drei Jahren (HR 0,60; p = 0,019). Die Ergebnisse waren in verschiedenen untersuchten Subgruppen vergleichbar. Zwei Patienten im Hochdosis-Arm und einer im Chemotherapie-Arm verstarben an Toxizitäten; insgesamt litten im Hochdosis-Arm mehr Patienten an schweren akuten Toxizitäten, aber diese traten nur nach dem einen Zyklus Busulfan/Melphalan auf, während schwere Toxizitäten im Kontrollarm sich nach mehreren Zyklen zeigen konnten. Zur Langzeit-Toxizität gibt es noch keine Daten.
Die Hochdosistherapie, die bei soliden Tumoren zuletzt kaum mehr eine Rolle zu spielen schien, ist Whelan zufolge damit als Standard bei diesen Patienten mit lokalisiertem Ewing-Sarkom anzusehen, die schlecht auf die Chemotherapie ansprechen oder große Tumoren aufweisen – sofern sie keine Kontraindikationen gegen Busulfan/Melphalan aufweisen.
Literatur
1. Chi Y et al. ASCO 21016, Abstract #11005.
2. ClincalTrials.gov NCT02449343.
3. Schöffski P et al. Lancet 2016; 387: 1629-37.
4. Hudgens S et al. ASCO 2016, Abstract #11015.
5. van der Graaf WT et al. Lancet 2012; 379: 1879-86.
6. Plummer R et al. Cancer Chemother and Pharmacol 2013; 71: 93-101.
7. Schmoll HJ et al. ASCO 2016, Abstract #11004.
8. Penel N et al. ASCO 2016, Abstract #11003.
9. Whelan J et al. ASCO 2016, Abstract #11000.

Prof. Dr. med. Clemens-Martin Wendtner
Klinik für Hämatologie, Onkologie,
Immunologie, Palliativmedizin, Infektiologie und Tropenmedizin, Klinikum Schwabing
Akademisches Lehrkrankenhaus der
Universität München
Kölner Platz 1, 80804 München
+49 89 3068-2228
+49 89 3068-3912
per E-Mail kontaktieren