Differenzialdiagnose myeloproliferativer Neoplasien
Hans Michael Kvasnicka, Martin Griesshammer
Differenzialdiagnostische molekulare und pathologische Betrachtung der MPN
Die BCR-ABL1-negativen myeloproliferativen Neoplasien (MPN) umfassen als hauptsächliche Subtypen neben der primären Myelofibrose (PMF) die essenzielle Thrombozythämie (ET) sowie die Polycythaemia vera (PV). Allen MPN ist neben einer klonalen Evolution ein signifikant unterschiedlicher Krankheitsverlauf gemeinsam [1, 2], der durch Komplikationen wie Thrombozythämie sowie (sekundäre) Myelofibrose und schließlich eine instabile Phase mit Akzeleration und nachfolgender Blastenkrise geprägt sein kann [3]. Die in den letzten Jahren entwickelten neuen Behandlungsmöglichkeiten [4] und die erheblichen Unterschiede hinsichtlich des Überlebens zwischen den einzelnen Subtypen erfordern nicht nur eine sichere Abgrenzung von anderen reaktiven oder hämatopoetischen Neoplasien [5], sondern auch eine eindeutige Klassifikation der einzelnen MPN-Subtypen [6–8]. Aus diesem Grunde wird ein synoptisches Vorgehen notwendig, welches neben den entsprechenden klinischen Daten sowie insbesondere molekulargenetischen Markern eine Knochenmarks-Morphologie als gleichwertige und unverzichtbare diagnostische Stütze umfasst [9].
Dieses Konzept kommt in der aktuellen sowie vor allem der kommenden WHO-Klassifikation zum Ausdruck [10], insbesondere vor dem Hintergrund, dass sich die einzelnen Subtypen der MPN durch ein sehr unterschiedliches Erscheinungsbild des Knochenmarks auszeichnen. Dieses gilt nicht nur für die Zytologie, sondern vor allem auch für die Histologie, da hier die Verhältnisse in situ (Histotopographie, quantitative Zuordnung der einzelnen Zellreihen, Faservermehrung) besser als im Ausstrichpräparat erkennbar sind [7, 8].
Im Gegensatz zur chronischen myeloischen Leukämie, die durch das Auftreten eines Philadelphia-Chromosoms und des sich hieraus ableitenden BCR-ABL1-Fusionsproduktes eindeutig molekular charakterisiert ist, weisen die übrigen Subtypen keine einheitliche genetische Abnormalität auf und zeigen besonders zu Beginn der Erkrankung oft ähnliche Befunde. In den letzten Jahren ist jedoch durch die Entdeckung wichtiger Mutationen (JAK2, CALR, MPL) ein wesentlicher Schritt für die molekulare Charakterisierung und auch Diagnose dieser Subtypen erreicht worden [10–12]. In diesem Zusammenhang ist aber festzuhalten, dass diese molekularen Befunde die immer noch kontrovers diskutierte Abgrenzung zwischen den häufig vorkommenden thrombozythämischen Frühformen der MPN in der Regel nicht ermöglichen [10].
Weiterhin ist anzumerken, dass die bei den einzelnen Subtypen anzutreffenden, unterschiedlich ausgeprägten histologischen Veränderungen häufig mit entsprechenden klinischen Befunden verknüpft sind, die wesentliche Aussagen zum stadienhaften Verlauf des Krankheitsgeschehens und daher zur Prognose zulassen [13, 14].
WHO Kriterien für die Diagnose der BCR-ABL1-negativen MPN
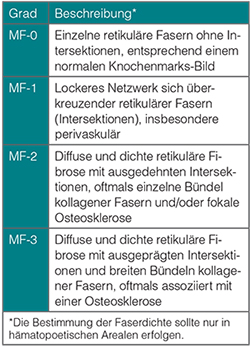
Im Gegensatz zu den von der PV-Study Group (PVSG) entwickelten Diagnosekriterien [13], welche die histopathologischen und neuen molekulargenetischen Parameter nicht berücksichtigen, stellen die zuletzt 2008 ergänzten und demnächst in einer aktualisierten Version erscheinenden Diagnosekriterien der WHO eine wesentliche Bereicherung für die Diagnosestellung der MPN dar [1, 15, 16]. Es werden sowohl die im Blut und Knochenmark prädominanten Zelllinien berücksichtigt als auch histopathologische Parameter miteinbezogen. Die Tabellen 1, 2 und 3 zeigen die derzeit gültigen WHO-Kriterien für die PMF, ET und PV, weiterhin wird in Tabelle 4 die zugrunde liegende semiquantitative Graduierung der Knochenmarks-Fibrose bei der PMF beschreiben [1, 17].
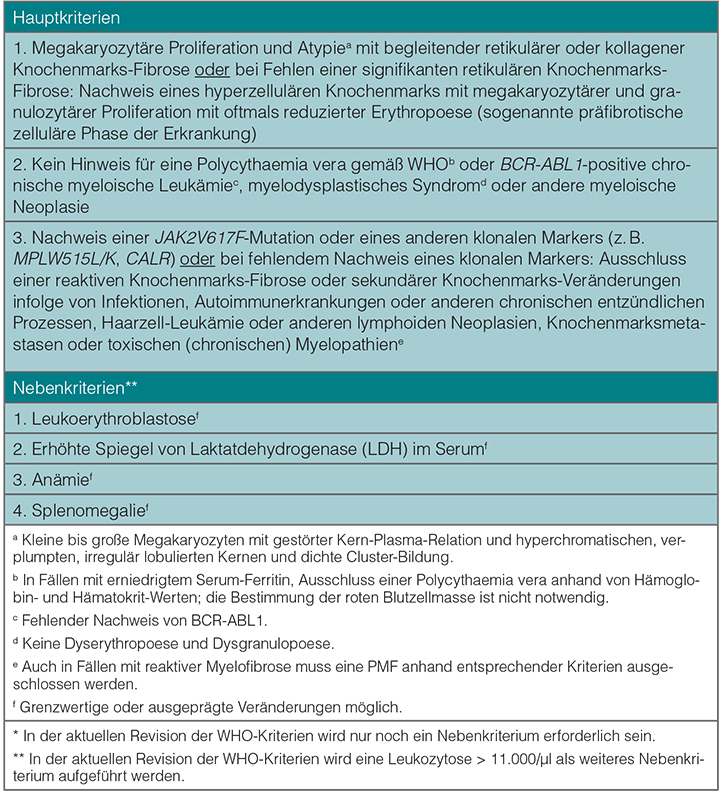


Im Hinblick auf die anstehende Aktualisierung der WHO-Kriterien ist anzumerken, dass für die PV die Hämoglobin- (Hb) und Hämatokrit(Hkt)-Schwellenwerte signifikant erniedrigt werden (Männer: Hb > 16,5 g/dl oder Hkt > 49%; Frauen: Hb > 16,0 g/dl oder Hkt >48%), um insbesondere auch frühe Formen der Erkrankung diagnostisch zu erfassen [18, 19]. Weiterhin wird in Analogie zur ET und PMF die Knochenmarks-Morphologie als Hauptkriterium aufgeführt werden, wobei jedoch in Fällen mit klinisch eindeutiger Erythrozytose (Männer: Hb > 18,5 g/dl, Hkt > 55,5%; Frauen: Hb > 16,5 g/dl, Hkt > 49,5%) und nachgewiesener JAK2-Mutation auf eine Knochenmarks-Biopsie verzichtet werden kann. Bei PMF wird vorgeschlagen, den Terminus Prä-PMF für initiale, präfibrotische Stadien der PMF einzuführen. Da die 2008 aufgestellten Nebenkriterien für die Diagnose der PMF (vgl. Tabelle 1) oftmals in den frühen Stadien nicht erfüllt werden, fordert die kommende Revision nur noch ein Nebenkriterium für die Etablierung der Diagnose, wobei jedoch zusätzlich eine Leukozytose > 11.000/µl als Nebenkriterium aufgenommen werden wird [10].
In Tabelle 5 wird auch die aktuelle Definition der nicht weiter klassifizierbaren Fälle dargestellt, die im Rahmen einer referenzpathologischen Begutachtung einen Anteil von deutlich weniger als 10% einnehmen [7, 8]. In dieser Gruppe müssen Effekte einer bereits vorangegangenen zytoreduktiven Therapie, welche das morphologische Bild des Knochenmarkes erheblich verändern kann, definitiv ausgeschlossen werden.

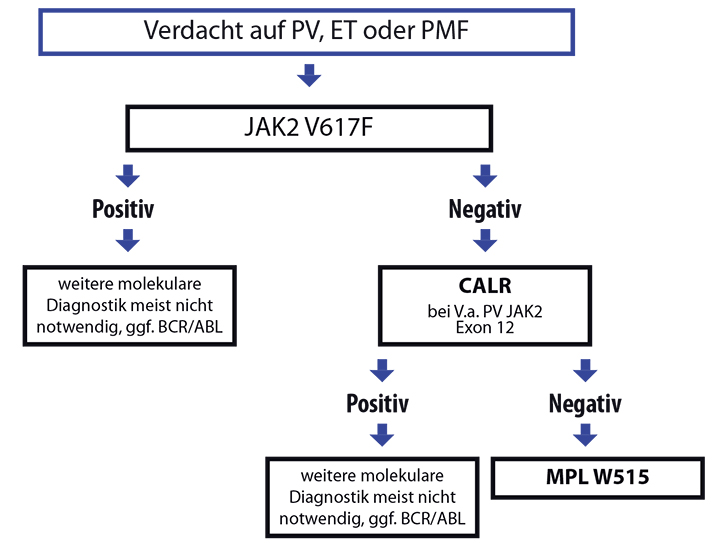
Durch Nachweis einer Mutation von JAK2, CALR oder MPL steht bei der Mehrzahl der MPN-Patienten ein molekularbiologischer Marker für die Diagnostik zur Verfügung (Tabelle 6). In der Regel wird bei klinischem Verdacht auf eine MPN zunächst ein Screening auf eine JAK2V617F-Mutation durchgeführt (Abb. 1). Sollte das Ergebnis negativ sein, wird in einem zweiten Schritt auf eine Mutation im Calreticulin-Gen (CALR) getestet, und nur wenn diese auch negativ ist, erfolgt ein abschließendes Screening auf eine mögliche MPLW515-Mutation [10–12]. Letztere tritt in 8% aller PMF-Fälle und in weniger als 5% der ET-Fälle auf. Bei JAK2/MPL-negativen PMF-Patienten liegt in 88% der Fälle eine Mutation im Calreticulin-Gen vor, insgesamt tritt in ca. 25% der PMF-Fälle eine CALR-Mutation auf. Die BCR-ABL-Genfusion wird in der Regel nur bestimmt, wenn eine CML als mögliche Differenzialdiagnose infrage kommt oder falls CALR-, JAK2V617F- und MPLW515-Mutation nicht gefunden werden. Der Algorithmus der stufenweisen molekularen Testung ist in Abb. 1 dargestellt.
Stellenwert der Knochenmarks-Biopsie
Die WHO-Kriterien ermöglichen eine bessere Differenzialdiagnose der BCR-ABL1-negativen MPN nach klinisch-pathologischen Kriterien. Eine besondere diagnostische Wertigkeit hat die Knochenmarks-Biopsie bei der Differenzierung der MPN mit Thrombozythämie, insbesondere ET und frühe PMF [7, 8, 13, 20–24]. Knochenmarks-Biopsien sollten trotz der Belastung für den Patienten nicht nur zur Festlegung der richtigen Diagnose (vor jeder Therapie), sondern auch zur regelmäßigen Kontrolle der MPN im Verlauf durchgeführt werden. Die histopathologische Untersuchung kann sowohl bei der Prognosestellung helfen, als auch die Entwicklung von Komplikationen wie beispielsweise die Progression zur Myelofibrose frühzeitig diagnostizieren. Der Vergleich zwischen PVSG- und WHO-Kriterien ergibt auch bei der Risikoberechnung der myelofibrotischen Transformation bei Patienten mit ET und PMF große Differenzen. Während die PVSG-Kriterien bei 27,5% der untersuchten ET-Patienten eine myelofibrotische Transformation prognostizieren, treten bei der nach WHO-Kriterien diagnostizierten ET nur sehr selten entsprechende Transformationen auf [25].
Primäre Myelofibrose
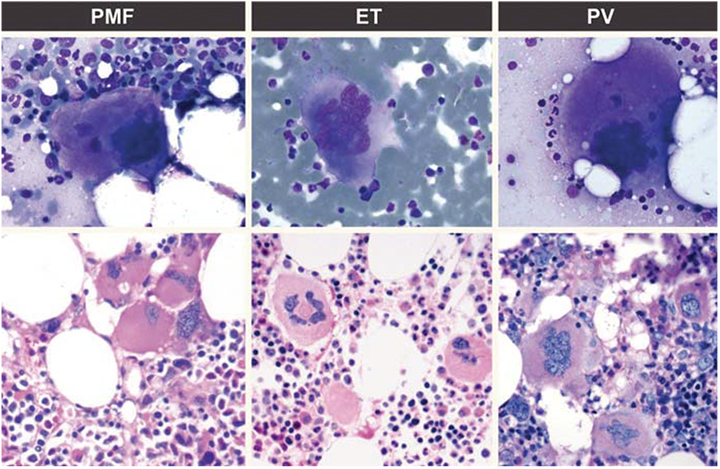
Die PMF zeichnet sich in den Spätstadien durch eine ausgeprägte kollagene Fibrose des Knochenmarks und eine extramedulläre Hämatopoese aus [26]. Alle drei hämatopoetischen Zelllinien können proliferieren. Die Erkrankung verläuft jedoch langsam fortschreitend und beginnt mit einem präfibrotischen Stadium, das durch ein hyperzelluläres Knochenmark ohne nennenswerte Knochenmarks-Fibrose charakterisiert ist [14, 21]. Im fibrotischen Stadium zeigt sich eine ausgeprägte retikuläre und kollagenöse Fibrose bis hin zur Osteomyelosklerose [27]. In diesem Stadium finden sich im leukoerythroblastischen Blutausstrich typischerweise tränenförmige Erythrozyten (Poikilozytose). Die Morphologie der Megakaryozyten ist für die Diagnose der PMF entscheidend, im Gegensatz zur ET oder PV ergibt sich hier eine eindeutige Reifungsstörung dieser Zelllinie, auch in den frühen Krankheitsstadien (Abbildung 2). Die Zellkerne sind meist verplumpt und hyposegmentiert, weiterhin wird eine atypische Nesterbildung, oftmals auch in endostaler Dislokation beobachtet [28, 29].
Essenzielle Thrombozythämie
Bei der ET steht eine konstante Thrombozythämie mit Werten über 450 x 109/l im Vordergrund, die auf eine ausgeprägte klonale Proliferation großer Megakaryozyten zurückzuführen ist [7, 8], die allerdings neben den überwiegend riesigen Zellformen eine insgesamt regelhafte Kern-Zytoplasma-Relation erkennen lassen und relativ locker im Knochenmark verteilt sind (Abbildung 2). Somit besteht ein auffallender Gegensatz zu den deutlichen histotopographischen und zytologischen Atypien der Megakaryozyten in den präfibrotischen Stadien der PMF. Granulo- und Erythropoese weisen dagegen im Vergleich zu den übrigen oben erwähnten Subtypen der MPN keine ausgeprägte Proliferation auf, und der Anteil primär diagnostizierter Fälle mit beginnender retikulärer Faservermehrung ist als verschwindend gering anzusehen [9, 25].
Polycythaemia vera
Die PV ist gekennzeichnet durch eine Erythropoetin(EPO)-unabhängige massive Vermehrung erythropoetischer Zellen. Die Zahl der Erythrozyten liegt bei > 6,5 x 1012/l, die arterielle Sauerstoffsättigung ist normal. Auch die Granulo- und Megakaryopoese kann gesteigert sein. In der frühen proliferativen, polyzythämischen Phase ist die Erythrozytenmasse signifikant erhöht [30]. In der späteren Phase zeigen sich eher Zytopenien mit Anämie, eine Knochenmarksfibrose und teils deutliche Hypersplenie als Ausdruck einer extramedullären Hämatopoese [28, 29]. Neben dieser post-polyzythämischen myeloischen Metaplasie mit Myelofibrose (post-PV-MF) zeigt sich im natürlichen Verlauf der PV selten auch eine Transformation zu einer akuten Leukämie. Die Ursache der PV ist unklar, es zeigt sich jedoch eine familiäre Häufung. Auch ionisierende Strahlung, toxische Substanzen und Viren werden bei einigen Patienten als Ursache diskutiert [30]. Histomorphologisch findet sich bei der PV ein hyperzelluläres Knochenmark mit trilinearer Proliferation (Panmyelose) [31]. Die Megakaryozyten sind im Gegensatz zur ET durch eine ausgeprägte Pleomorphie gekennzeichnet (Abbildung 2), d. h. es finden sich kleine, mittelgroße und riesengroße Zellformen zumeist in lockerer interstitieller Verteilung. Eine Reifungsstörung dieser Zelllinie ist nicht erkennbar.
Literatur
1. Swerdlow S et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues Lyon, France: IARC Press; 2008.
2. Tefferi A et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: Recommendations from an ad hoc international expert panel. Blood 2007; 110: 1092-7.
3. Tefferi A et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014; 124: 2507-13; quiz: 2615.
4. Barbui T et al. Philadelphia-negative classical myeloproliferative neoplasms: Critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011; 29: 761-70.
5. Tefferi A et al. Myeloproliferative neoplasms: Contemporary diagnosis using histology and genetics. Nat Rev Clin Oncol 2009; 6: 627-37.
6. Kvasnicka HM, Thiele J. The impact of clinicopathological studies on staging and survival in essential thrombocythemia, chronic idiopathic myelofibrosis, and polycythemia rubra vera. Semin Thromb Hemost 2006; 32: 362-71.
7. Kvasnicka HM, Thiele J. Classification of Ph-negative chronic myeloproliferative disorders--morphology as the yardstick of classification. Pathobiology 2007; 74: 63-71.
8. Kvasnicka HM, Thiele J. Prodromal myeloproliferative neoplasms: The 2008 WHO classification. Am J Hematol 2010; 85: 62-9.
9. Barbui T et al. Problems and pitfalls regarding WHO-defined diagnosis of early/prefibrotic primary myelofibrosis versus essential thrombocythemia. Leukemia 2013; 27: 1953-8.
10. Tefferi A et al. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia 2014; 28: 1407-13.
11. Klampfl T et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013; 369: 2379-90.
12. Nangalia J et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013; 369: 2391-405.
13. Thiele J, Kvasnicka HM. Chronic myeloproliferative disorders with thrombocythemia: A comparative study of two classification systems (PVSG, WHO) on 839 patients. Ann Hematol 2003; 82: 148-52.
14. Thiele J, Kvasnicka HM. Grade of bone marrow fibrosis is associated with relevant hematological findings - a clinicopathological study on 865 patients with chronic idiopathic myelofibrosis. Ann Hematol 2006; 85: 226-32.
15. Tefferi A et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014; 28: 2300-3.
16. Vardiman JW et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009; 114: 937-51.
17. Thiele J et al. European consensus for grading of bone marrow fibrosis and assessment of cellularity. Haematologica 2005; 90: 1128-32.
18. Barbui T et al. Masked polycythemia vera diagnosed according to WHO and BCSH classification. Am J Hematol 2014; 89: 199-202.
19. Barbui T et al. Masked polycythemia vera (mPV): Results of an international study. Am J Hematol 2014; 89: 52-4.
20. Thiele J, Kvasnicka HM. Diagnostic differentiation of essential thrombocythaemia from thrombocythaemias associated with chronic idiopathic myelofibrosis by discriminate analysis of bone marrow features - a clinicopathological study on 272 patients. Histol Histopathol 2003; 18: 93-102.
21. Thiele J, Kvasnicka HM. Prefibrotic chronic idiopathic myelofibrosis - a diagnostic enigma? Acta Haematol 2004; 111: 155-9.
22. Thiele J, Kvasnicka HM. Hematopathologic findings in chronic idiopathic myelofibrosis. Semin Oncol 2005: 32: 380-94.
23. Thiele J, Kvasnicka HM. Clinicopathological criteria for differential diagnosis of thrombocythemias in various myeloproliferative disorders. Semin Thromb Hemost 2006; 32: 219-30.
24. Thiele J, Kvasnicka HM. The 2008 WHO diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis. Curr Hematol Malig Rep 2009; 4: 33-40.
25. Barbui T et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: An international study of 1,104 patients. J Clin Oncol 2011; 29: 3179-84.
26. Barosi G. Myelofibrosis with myeloid metaplasia: Diagnostic definition and prognostic classification for clinical studies and treatment guidelines. J Clin Oncol 1999; 17: 2954-70.
27. Thiele J, Kvasnicka HM. Myelofibrosis - what's in a name? Consensus on definition and EUMNET grading. Pathobiology 2007; 74: 89-96.
28. Thiele J et al. Bone marrow histopathology in myeloproliferative disorders - current diagnostic approach. Semin Hematol 2005; 42: 184-95.
29. Thiele J et al. Bone marrow histopathology in the diagnosis of chronic myeloproliferative disorders: A forgotten pearl. Best Pract Res Clin Haematol 2006; 19: 413-37.
30. Spivak JL. Polycythemia vera: Myths, mechanisms, and management. Blood 2002; 100: 4272-90.
31. Thiele J, Kvasnicka HM. Diagnostic impact of bone marrow histopathology in polycythemia vera (PV). Histol Histopathol 2005; 20: 317-28.

Prof. Dr. Hans Michael Kvasnicka
Senckenberg Institut für Pathologie,
Universität Frankfurt, Frankfurt

Prof. Dr. Martin Griesshammer
Klinik für Hämatologie, Onkologie und
Palliativmedizin, Mühlenkreiskliniken,
Johannes Wesling Klinikum Minden, Minden