Morbus Waldenström
Alexander Grunenberg, Christian Buske
Epidemiologie
Die Erkrankung macht etwa 1–2% aller hämatologischen Neoplasien aus. Betroffen sind in der Regel vornehmlich männliche Patienten höheren Alters mit einem mittleren Erkrankungsalter von 63–68 Jahren [1]. Epidemiologische Daten beschreiben eine Häufigkeit von 1.500 Neuerkrankungen pro Jahr in den USA. Eine familiäre Häufung ist beim Morbus Waldenström erkennbar [2]. Bei bis zu 20% der Betroffenen weist ein Verwandter ersten Grades eine B-Zell-Neoplasie auf [3, 4].
Genetische Veränderungen
Die häufigste zytogenetische Aberration ist die Deletion des langen Arms von Chromosom 6 (6q-Deletion): Sie ist bei bis zu 50% der Patienten mit Morbus Waldenström nachweisbar und mit einer ungünstigen Prognose assoziiert. Eine 6q-Deletion ist nicht spezifisch für den Morbus Waldenström, sondern findet sich bei einer Reihe anderer indolenter Lymphome wie auch beim multiplen Myelom. Weitaus spezifischer ist eine Trisomie 4, die aber wiederum nur bei bis zu 20% der Patienten auftritt.
Die für die Diagnostik derzeit bedeutendste genetische Alteration ist die MYD88-L265P-Mutation, die bei ca. 90% der Patienten mit Morbus Waldenström vorkommt. MYD88 ist physiologisch an der angeborenen und erworbenen Immunantwort beteiligt. Auf zellulärer Ebene wird es durch Bindung von Liganden an Toll-like- oder Interleukin-Rezeptoren aktiviert und dient als Aktivator des JAK-STAT- und NF-κB-Signalweges. Experimentellen Daten zufolge führt eine Mutation im MYD88-Gen zu einer konstitutiven Aktivierung der oben genannten Signalkaskaden und wirkt dadurch anti-apoptotisch [5, 6].
Eine weitere bedeutende genetische Alteration betrifft das CXCR4-Gen; die Bindung von dessen Proteinprodukt an CXCL-12 ist für die Bindung der Lymphomzellen an das Mikromilieu im Knochenmark von eminenter Bedeutung. 30% der Patienten weisen aktivierende Mutationen in diesem Gen auf. Bei Betrachtung des Mutationsstatus des MYD88- und des CXCR4-Gens können beim Morbus Waldenström drei genetische Gruppen voneinander unterschieden werden: Die Gruppe mit Mutationen in beiden Genen, die Gruppe mit MYD88-Mutation und CXCR4-Wildtyp sowie die Gruppe ohne jegliche Mutation in beiden Genen. Interessanterweise scheint CXCR4 bei Vorliegen des MYD88-Wildtyps nicht mutiert zu sein. Diese Unterteilung in genetische Subgruppen erscheint klinisch bedeutsam, da Patienten mit CXCR4-Mutation schlechter auf Ibrutinib anzusprechen scheinen und Patienten mit dem Wildtyp beider Gene überraschenderweise die geringsten Ansprechraten zeigen [7, 8].
Klinik
Krankheitssymptome werden durch die Knochenmarkinfiltration der Lymphomzellen und durch die zum Teil hohen IgM-Spiegel verursacht. Folge der Ausdehnung der Lymphomzellen im Knochenmark ist eine Verdrängung der physiologischen Hämatopoese mit Anämie (70%), Leukopenie (10%) und Thrombopenie (30%). Die Betroffenen klagen neben der typischen B-Symptomatik (Gewichtsverlust, Nachtschweiß, Fieber) über Müdigkeit (85%), haben ein erhöhtes Infektionsrisiko und weisen eine erhöhte Blutungsneigung auf. Ein Teil der Patienten entwickelt eine Lymphadenopathie (40%) und/oder Hepatosplenomegalie (30%).
Ein anderer Teil der Symptome ist IgM-assoziiert. Die Folge einer hohen IgM-Konzentration ist ein Hyperviskositäts-Syndrom. Es kommt zu Mikrozirkulationsstörungen mit Raynaud Symptomatik (5%) und neurologischen Störungen (20%). Die Patienten klagen über Schwindel, Kopf- und Sehstörungen, Bewusstseinsstörungen und periphere Neuropathien (z. B. Gangstörungen). Selten kommen Leichtketten-Amyloidosen, Kryoglobulinämien oder IgM-Kälteagglutinine mit entsprechender klinischer Symptomatik vor [9].
Differenzialdiagnosen
Es gibt eine Reihe von Erkrankungen, die mit einer IgM-Gammopathie assoziiert sind. Zu nennen sind hier neben dem Morbus Waldenström das IgM-MGUS (monoklonale Gammopathie unklarer Signifikanz), andere Non-Hodgkin-Lymphome, das IgM-Myelom, die Amyloid-Leichtketten-Amyloidose (AL-Amyloidose) sowie Paraprotein-assoziierte Erkrankungen wie beispielsweise die Polyneuropathie oder Kryoglobulinämie.
Diagnostik
Nach der aktuellen WHO-Klassifikation von 2008 für hämatologische Neoplasien ist für die Diagnose des Morbus Waldenström der histopathologische Nachweis eines lymphoplasmozytischen Lymphoms im Knochenmark (≥ 10%) mit monoklonaler IgM-Gammopathie unabhängig von der IgM-Serumkonzentration zu fordern [10, 11]. Zum initialen Staging und zur Risikoklassifikation ist eine ausführliche Diagnostik essenziell. Hierzu zählen:
Anamnese (B-Symptomatik) und körperliche Untersuchung, Blutbild einschließlich Differenzialblutbild und Retikulozytenbestimmung, Blutsenkungsgeschwindigkeit und Gesamteiweiß, Albumin, Serum-Elektrophorese sowie die quantitative Bestimmung der Immunglobuline und der Nachweis des monoklonalen IgM-Proteins durch Immunfixation. Weitere zu bestimmende laborchemische Parameter sind AST, ALT, AP, γ-GT, Bilirubin, glomeruläre Filtrationsrate, Kreatinin, Harnsäure, Blutzucker, LDH, β2-Mikroglobulin und die globalen Gerinnungsparameter Quick-Wert und partielle Thromboplastin-Zeit (PTT). Kälteagglutinine und Kryoglobuline können die quantitative Bestimmung des IgM mit beeinflussen und sollten daher mitbestimmt werden. Im Falle einer peripheren Neuropathie sollten ferner folgende Antikörper bestimmt werden: Anti-MAG (Myelin-assoziiertes Glykoprotein), Anti-Gangliosid M1 und Anti-Sulfatid-IgM-Antikörper. Bei Erstdiagnose sollte eine Thorax-Röntgenaufnahme in zwei Ebenen, eine Sonografie von Hals und Oberbauch sowie ein CT von Thorax und Abdomen durchgeführt werden. Die PET-Diagnostik ist beim
M. Waldenström nicht indiziert.
In der Knochenmark-Zytologie und -Histologie zeigt sich morphologisch eine Infiltration durch Lymphozyten mit plasmozytoider beziehungsweise Plasmazell-Differenzierung. In der FACS-Analyse (Durchflusszytometrie) exprimieren die Lymphomzellen typischerweise die Oberflächenmarker CD19, CD20, CD22 sowie CD79a, und in 80–90% der Fälle fehlt die Expression von CD5, CD10 und CD23. Das Fehlen der letztgenannten Marker hilft häufig bei der differenzialdiagnostischen Abgrenzung zum follikulären Lymphom, zur CLL und zum Mantelzell-Lymphom. Jedoch ist anzumerken, dass eine Expression der Marker CD5, CD10 und CD23 keine Ausschlussdiagnose für den Morbus Waldenström ist [12]. Diagnostische Bedeutung hat die Bestimmung der MYD88-Mutation, die zwar nicht spezifisch für den Morbus Waldenström ist und hier auch in 10% der Fälle fehlt, die aber bei diagnostisch unklaren Fällen z. B. die Abgrenzung vom multiplen Myelom, das keine MYD88-Mutationen aufweist, erleichtern kann.
Stadium
Aufgrund des obligaten Knochenmark-Befalls bedeutet die Diagnose eines Morbus Waldenström per definitionem ein fortgeschrittenes Stadium.
Risiko- und Prognoseklassifikation
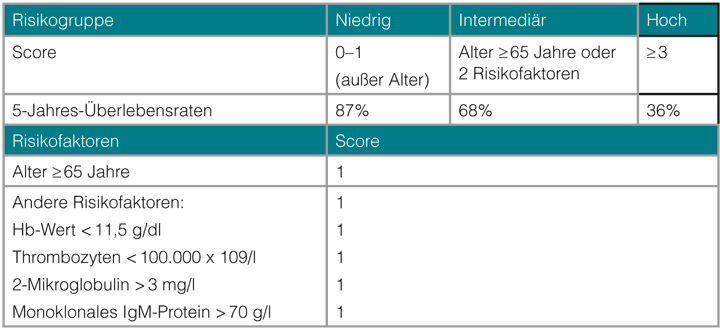
Neu diagnostizierte Patienten sollten bei Diagnosestellung zur Hilfe bei der Therapieentscheidung anhand des seit 2009 speziell für den M. Waldenström entwickelten internationalen prognostischen Score (ISSWM) klassifiziert werden. Innerhalb dieses Scores werden drei Risikogruppen unterschieden, mit 5-Jahres-Überlebensraten von 86% für die Niedrigrisiko-Gruppe bis hin zu 36% für die Hochrisiko-Gruppe (s. Tabelle 1; [13]).
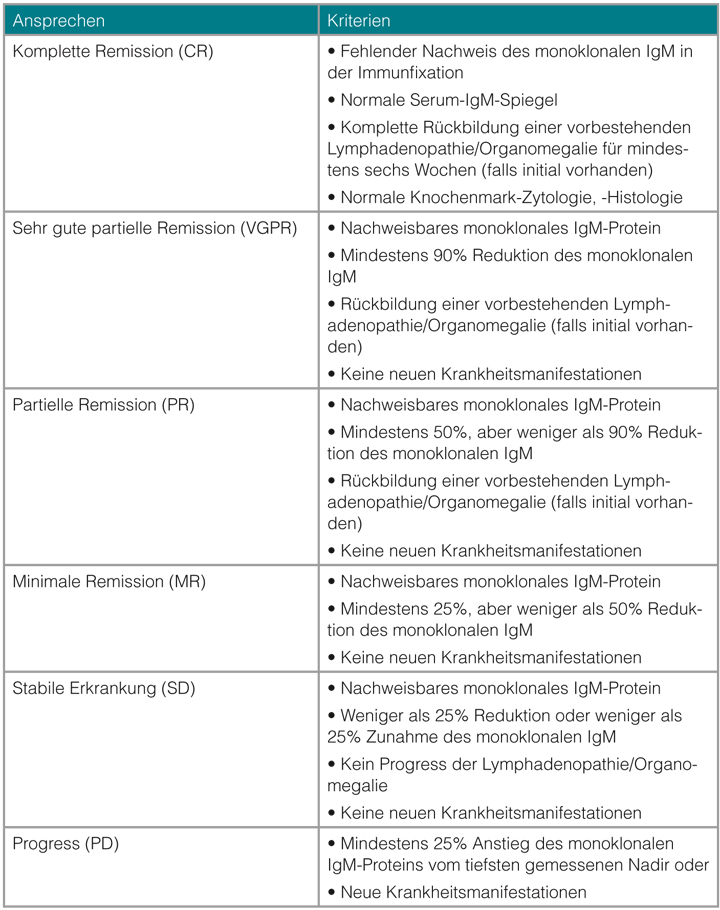
Remissionsstatus
Zur Beurteilung des Remissionsstatus existiert ein international anerkannter Konsensus [14]. Besondere Bedeutung hat hierbei die Konzentration des IgM-Paraproteins. Sein Verschwinden ist Voraussetzung für die Definition einer kompletten Remission (Tabelle 2). Aufgrund der Variabilität der Kinetik der IgM-Reduktion bei verschiedenen Therapiekonzepten und der oft bestehenden Diskrepanz zwischen der IgM-Konzentration und dem Knochenmarkansprechen sollte neben der IgM-Messung auch auf regelmäßige Knochenmark-Punktionen geachtet werden.
Therapie
Aktuell gibt es keinen Goldstandard in der Behandlung des Morbus Waldenström. Das ist zum einen der bisher verhältnismäßig überschaubaren Anzahl randomisierter klinischer Studien geschuldet, zum anderen auch der Tatsache, dass in der Behandlung stets die individuellen Patienten-Charakteristika Berücksichtigung finden müssen. Da der Morbus Waldenström eine Erkrankung des höheren Lebensalters ist, ist mehr als die Hälfte der Patienten bei Diagnosestellung über 70 Jahre alt und hat in nicht unerheblichem Maße entsprechende Komorbiditäten, sodass häufig nur weniger intensive Therapieregimes in Betracht kommen. Dem stehen jüngere Patienten mit aggressiverem Erkrankungsverlauf gegenüber, die durchaus für intensivere Therapien infrage kommen sollten.
Erstlinientherapie
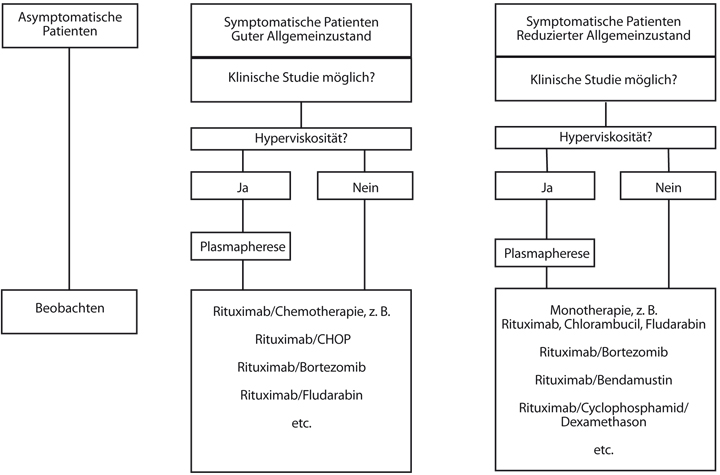
Asymptomatische Patienten
Vergleichbar anderen indolenten Lymphomen im fortgeschrittenen Stadium wird für asymptomatische Patienten eine „Watch & Wait“-Strategie empfohlen. Nur Patienten mit Lymphom-assoziierten Symptomen sollen einer Therapie zugeführt werden [15, 16]. Im Falle des Morbus Waldenström zählen hierzu: Hämoglobin-Spiegel unter
10 g/dl oder Thrombozyten-Konzentrationen unter 100.000/µl, ausgeprägte Hepatosplenomegalie und Lymphadenopathie, IgM-assoziierte Symptome wie das Hyperviskositäts-Syndrom, eine Amyloidose, eine symptomatische Kryoglobulinämie, Kälteagglutinin-Krankheit, Neuropathie. Der Nachweis eines monoklonalen IgM ist per se keine Therapieindikation [16].
Symptomatische Patienten in gutem Allgemeinzustand
Als einer der Standards in der Behandlung des Morbus Waldenström für medizinisch fitte Patienten gilt die Kombination von Rituximab mit einer Chemotherapie. Weitere Therapieoptionen sind die Rituximab-Monotherapie, Bortezomib in Monotherapie oder in Kombination mit Rituximab.
Rituximab-Monotherapie
Die Rituximab Monotherapie ist bei Morbus Waldenström weniger effektiv als beim follikulären Lymphom. Viermalige Gaben in wöchentlichem Abstand führen zu Ansprechraten von 20–30%. Wird die Rituximab-Therapie im Anschluss fortgeführt, erhöht sich die Rate auf bis zu 50% [17]. Problematisch ist das verzögerte Ansprechen unter Rituximab-Monotherapie insbesondere bei Patienten mit Symptomen eines Hyperviskositäts-Syndroms oder hohen IgM-Spiegeln.
Unter der anfänglich begonnenen Therapie mit Rituximab kann oftmals ein sogenanntes IgM-Flare-Phänomen auftreten, d. h. ein vorübergehender Anstieg der IgM-Serumspiegel [18]. Bei IgM-Spiegeln > 50 g/dl oder einer Serumviskosität von > 3,5 cp (Centipoise) besteht ein erhöhtes Risiko für ein Hyperviskositäts-Syndrom, weshalb zunächst eine Plasmapherese durchgeführt werden und/oder zunächst bis zur Senkung des IgM-Spiegels auf die Gabe von Rituximab verzichtet werden sollte. Allerdings ist das IgM-Flare-Phänomen nicht gleichzusetzen mit einem Therapieversagen, und die meisten Patienten kehren innerhalb der ersten drei Monate nach Therapiestart wieder auf ihren IgM-Ausgangswert zurück.
Eine Rituximab-Erhaltungstherapie, die bereits beim follikulären Lymphom Anwendung findet, wird möglicherweise in naher Zukunft auch integraler Bestandteil der Behandlung des Morbus Waldenström sein. Leider gibt es derzeit keine prospektive Studie für Patienten mit Morbus Waldenström für diese Therapie. Es existieren jedoch Daten einer retrospektiven Studie mit 248 Rituximab-naiven Patienten, in der die Erhaltung zu einer Verbesserung des progressionsfreien wie auch des Gesamtüberlebens führte [19]. Die Überlegenheit einer Rituximab-Erhaltungstherapie muss dennoch erst in randomisierten Studien bestätigt werden, bevor sie außerhalb von Studien empfohlen werden kann.
Rituximab in Kombination mit Chemotherapie
Alkylanzien
In einer prospektiv randomisierten Studie der Deutschen Studiengruppe für Niedrigmaligne Lymphome wurden 48 Patienten in einen CHOP- (Cyclophosphamid, Doxorubicin, Vincristin, Prednison) und einen R-CHOP-Arm randomisiert [20]. Durch die Hinzunahme von Rituximab konnte ein signifikant höheres Gesamtansprechen erzielt werden (91% vs. 60% unter CHOP; p = 0,0188). Des Weiteren konnte in der Studie im R-CHOP-Arm eine signifikante Verlängerung der Zeit bis zum Therapieversagen beobachtet werden (im Median 63 vs. 22 Monate im CHOP-Arm; p = 0,0241).
Ähnlich gute Resultate wurden von der Eastern Cooperative Oncology Group (ECOG) berichtet: Hier erzielten 91% der Patienten nach median 1,6 Monaten eine partielle Remission [21]. Hauptnebenwirkungsrate der R-CHOP-Therapie ist die Myelosuppression, die wiederum für ältere Patienten in eingeschränktem Allgemeinzustand als zu toxisch angesehen wird. Jedoch ist für junge Patienten, bei denen eine mögliche Stammzell-Apherese mit nachfolgender myeloablativer Therapie in Betracht gezogen wird, dieses Therapieregime weiterhin eine Option.
Bendamustin
Das Chemotherapeutikum Bendamustin weist chemisch sowohl Eigenschaften eines Nukleosid-Analogons als auch eines Alkylans auf. Infolge seines geringen Toxizitätsprofils und seiner nachgewiesenen hervorragenden therapeutischen Effektivität bei der Behandlung des follikulären Lymphoms hat diese Substanz in den letzten Jahren wieder vermehrt Beachtung gefunden.
Eine Subgruppenanalyse einer großen randomisierten Studie verglich unter anderem bei 40 Patienten mit Morbus Waldenström eine Kombinationstherapie aus Bendamustin und Rituximab (BR) mit dem R-CHOP-Regime. In beiden Therapiearmen wurden hohe Ansprechraten von weit über 90% erreicht, jedoch waren beide Arme nicht in der Lage, komplette Remissionen zu erzielen [22]. Die Dauer des Ansprechens lag bei Patienten unter BR höher als im R-CHOP-Arm.
BR stellt somit ein weiteres mögliches Therapieregime insbesondere für die älteren Patienten mit Morbus Waldenström dar und wird von vielen als einer der Standards in der Behandlung dieser Erkrankung angesehen.
Purinnukleosid-Analoga
Eine weitere in der Therapie des Morbus Waldenström eingesetzte Substanzklasse sind die Nukleosid-Analoga Cladribin und Fludarabin. In kleineren Phase-II-Studien konnten bei therapienaiven Patienten mit Fludarabin Ansprechraten von 55–90% erreicht werden [23, 24]. In einer weiteren größeren Phase-II-Studie mit einem Kollektiv von 118 Patienten wurden Gesamtansprechraten von 38% erzielt mit 3% Komplettremissionen. Das mediane ereignisfreie und Gesamtüberleben lag bei drei und knapp sieben Jahren [25]. Hauptnebenwirkungen der Therapie mit Purin-Analoga sind Myelosuppression und T-Zell-Depletion. Hier ist eine Infektionsprophylaxe mit Cotrimoxazol essenziell.
Kombinationstherapien mit Nukleosid-Analoga wurden sowohl in der Erst- als auch in der Zweitlinientherapie untersucht. In einer Studie mit 29 Patienten, entweder therapienaiv oder im Rezidiv, wurde Cladribin mit Rituximab kombiniert und in insgesamt vier Zyklen verabreicht. Nach einer medianen Follow-up-Zeit von 43 Monaten lag die Gesamtansprechrate bei beachtlichen knapp 90% mit sieben Komplettremissionen, 16 partiellen Remissionen und drei „Minor Responses“ [26].
Die Kombination von Rituximab mit Fludarabin erbrachte eine Gesamtansprechrate von über 95% mit einer sehr guten partiellen Remission in mindestens 83% der Fälle. Eingeschlossen in diese Studie der WMCTG wurden ebenfalls therapienaive Patienten und Patienten im Rezidiv [27].
In einer 2012 von Tedeschi et. al publizierten Studie wurden behandlungsbedürftige Patienten (n = 43) mit sechs Zyklen aus Fludarabin, Cyclophosphamid und Rituximab (FCR) therapiert. Berichtet wurden in diesem Kollektiv Gesamtansprechraten von knapp 80%, darunter 11,6% Komplettremissionen, 20% sehr gute partielle und 41,8% partielle Remissionen. Allerdings zeigte sich eine hohe Rate an myelotoxischen Nebenwirkungen. Bei 35% der Patienten wurde eine lang, in Einzelfällen bis zu 15 Monate anhaltende Neutropenie beobachtet [28]. Aufgrund dieser ausgeprägten Myelosuppression wird daher nach derzeitiger Studienlage eine Therapie mit sechs Zyklen FCR nicht empfohlen.
Rituximab in Kombination mit Bortezomib
Neben einer Reihe von älteren Phase-II-Studien, in denen die Wirksamkeit von Bortezomib in Monotherapie nachgewiesen werden konnte, veröffentlichten Ghobrial et al. 2010 eine Studie, in der sie Rituximab in Kombination mit Bortezomib bei 26 bisher unbehandelten Patienten mit der Diagnose Morbus Waldenström untersuchten. Hierbei erreichten 58% der Patienten eine partielle und 8% eine komplette bzw. nahezu komplette Remission. Das ereignisfreie 1-Jahres-Überleben lag bei 79%, und es wurden keine höhergradigen Neuropathien beobachtet [29]. Dabei wurde Bortezomib einmal wöchentlich appliziert.
Symptomatische Patienten in reduziertem Allgemeinzustand
Wie bereits oben erwähnt, ist der Morbus Waldenström eine Erkrankung, die in der Regel ältere Patienten betrifft, die häufig an Komorbiditäten leiden und deshalb keine dosisintensiven Therapien tolerieren.
Eine mögliche und insbesondere für Patienten mit einem Hyperviskositäts-Syndrom anerkannte Therapieoption ist die Plasmapherese. Sie wird allgemein gut vertragen und vermeidet Nebenwirkungen, wie sie mit Chemotherapien assoziiert sind. Allerdings sollte, da die Wirksamkeit auch einer erfolgreichen Plasmapherese begrenzt ist, danach eine weitere Therapie angeschlossen werden. Dies kann z. B. die oben beschriebene Rituximab-Monotherapie sein.
Ein interessantes Regime wurde in einer Phase-II-Studie von Dimopoulos et al. vorgestellt, in der 72 bisher therapienaive Patienten unter Dexamethason, Rituximab und Cyclophosphamid (DRC) eine dokumentierte Ansprechrate von 83% mit 67% partiellen und 7% kompletten Remissionen erzielten. Bei sehr guter Verträglichkeit lag das progressionsfreie 2-Jahres-Überleben in dieser Kohorte bei 80%. Beispielsweise erlitten nur 9% der Patienten eine Neutropenie vom Grad 3 oder 4, Grad-3- oder Grad-4-Thrombozytopenien traten überhaupt nicht auf.
Insgesamt scheint somit dieses Regime für ältere Patienten eine vielversprechende Option darzustellen [30]. Es lässt sich auch hervorragend mit neueren Substanzen kombinieren. So testet derzeit das Europäische Konsortium für den Morbus Waldenström (ECWM; www.ecwm.eu) in einer prospektiv randomisierten Studie, ob die Wirksamkeit von DRC durch Hinzunahme von Bortezomib weiter gesteigert werden kann.
Rituximab in Kombination mit Bendamustin ist eine weitere Therapieoption für ältere Patienten und kann z. B. dosisreduziert (Bendamustin 70 mg/m2 an Tag 1 und 2 eines 28-Tage-Zyklus) appliziert werden.
Eine Kombinationstherapie aus Rituximab und Bortezomib ist prinzipiell auch bei komorbiden Patienten anwendbar; Bortezomib sollte dabei wöchentlich subkutan appliziert werden.
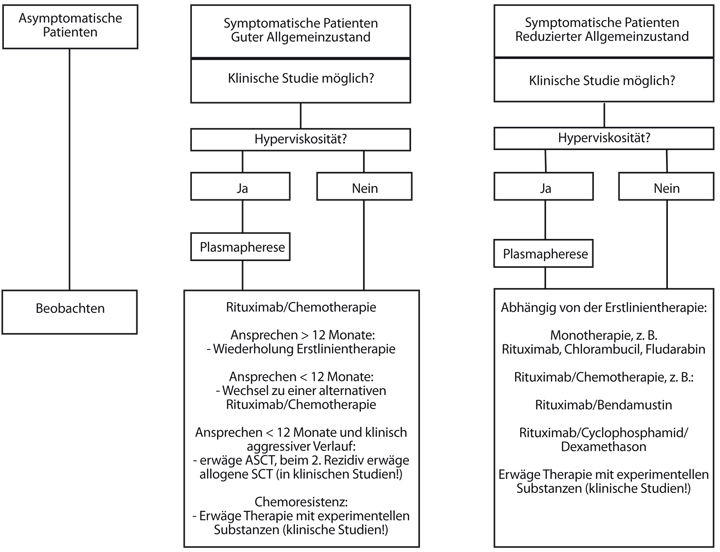
Rezidivtherapie beim Morbus Waldenström
Außerhalb von Studien ist die Kombination aus Rituximab und Chemotherapie in vielen Fällen immer noch das Rückgrat der Therapie des rezidivierten/refraktären M. Waldenström. Bei längerer Remissionsdauer nach initialer Behandlung können die Patienten optional erneut mit der identischen oder einer alternativen Rituximab-/Chemotherapie behandelt werden. Bei klinisch aggressivem Verlauf und jüngeren Patienten gilt in vielen Zentren die autologe Stammzelltransplantation (ASZT) als ein weiteres wichtiges Instrument in der Rezidivtherapie.
Rituximab-/Chemotherapie
Ähnlich wie in den Richtlinien zum follikulären Lymphom bildet die Chemotherapie in Kombination mit Rituximab das Rückgrat der Behandlung bei Patienten mit rezidiviertem M. Waldenström, sofern der Rückfall nicht innerhalb des ersten halben Jahres nach vorheriger Behandlung mit Rituximab auftritt. Es besteht Einverständnis, dass wechselnde Chemotherapien eingesetzt werden können, wenn der Rückfall innerhalb des ersten Jahres (Empfehlungen des International Workshop on Morbus Waldenström; [31]) oder der ersten beiden Jahre auftritt (mSMART: klinische Konsens-Empfehlungen der Mayo Clinic; [32]). Die Wahl der Chemotherapie hängt dabei von der vorherigen Behandlung ab: Wurde der Patient zunächst mit Rituximab und einem Alkylans behandelt, empfiehlt sich im Weiteren die Kombination von Rituximab mit Nukleosid-Analoga oder mit Bendamustin oder Bortezomib und umgekehrt. Außerdem muss berücksichtigt werden, ob eine autologe Stammzelltransplantation infrage kommt. In diesem Fall sollten Stammzell-toxische Therapien wie beispielsweise wiederholte Anwendungen von Nukleosid-Analoga vermieden werden.
Bei Rituximab–Refraktärität (Rezidiv innerhalb von sechs Monaten nach Gabe von Rituximab und Chemotherapie) ist Bendamustin als sehr wirksame Monotherapie zumindest beim follikulären Lymphom beschrieben worden [33]. Reagieren diese Patienten chemosensibel und kommen sie für eine autologe Stammzelltransplantation (ASZT) infrage, so stellt eine myeloablative Chemotherapie, gefolgt von einer autologen Stammzelltransplantation, in diesen klinisch aggressiven Fällen eine gute Option dar.
Autologe Stammzelltransplantation
Mehrere Berichte dokumentieren die Machbarkeit und Effektivität einer myeloablativen Chemotherapie, gefolgt von einer autologen Stammzelltransplantation [34]. Die umfassendste Analyse wurde von der European Group for Blood and Marrow Transplantation (EBMT) publiziert, die retrospektiv die Ergebnisse von 158 Patienten mit
M. Waldenström zwischen 1991 und 2005 untersuchte, die sich einer autologen Stammzelltransplantation unterzogen. Die überwiegende Mehrzahl der Patienten (93%) zeigte zum Zeitpunkt der Transplantation ein Ansprechen auf die Chemotherapie. Das progressionsfreie Überleben lag nach fünf Jahren bei 39,7%, das Gesamtüberleben bei 68,5%. Multivariate Analysen zeigten, dass eine chemoresistente Erkrankung bei ASZT und mindestens drei Vortherapien die wichtigsten unabhängigen prognostischen Faktoren für ein deutlich kürzeres progressionsfreies Überleben waren. Im Hinblick auf das Gesamtüberleben waren Faktoren wie drei oder mehr Behandlungslinien, Chemoresistenz bei ASZT, männliches Geschlecht und ein Alter > 50 Jahre prognostisch deutlich ungünstiger. Die ASZT wurde mit einer niedrigen nicht durch Rückfälle bedingten 1-Jahres-Mortalität von 3,8% gut toleriert. Das kumulative Risiko für sekundäre Tumoren betrug jedoch nach fünf Jahren 8,4% inklusive drei Patienten mit akuter myeloischer Leukämie (AML) und vier Patienten mit einem myelodysplastischen Syndrom (MDS; [35]).
Auf der Grundlage dieser Erfahrungen stellt eine Hochdosistherapie, gefolgt von ASZT, bei chemosensitivem rezidiviertem M. Waldenström eine wichtige Behandlungsoption für medizinisch fitte Patienten dar, insbesondere bei klinisch aggressiveren Fällen oder bei einem hohen IWMSS.
Allogene Stammzelltransplantation
Es gibt mehrere klinische Berichte, die zeigen, dass auch eine allogene Transplantation bei M. Waldenström durchführbar und wirksam ist. Die größte Untersuchung wurde wiederum von der EBMT durchgeführt, die über die langfristigen Ergebnisse bei Waldenström-Patienten berichtet [36]:
Die Patienten erhielten ein Transplantat entweder nach myeloablativer Konditionierung (n = 37) oder nach Konditionierung mit reduzierter Intensität (RIC; n = 49). 47 Patienten hatten drei oder mehr vorangegangene Therapien hinter sich. Die Rückfallrate nach drei Jahren lag bei 11% nach myeloablativer Konditionierung und bei 25% nach RIC. Progressionsfreie und Gesamtüberlebensrate nach fünf Jahren betrugen für die myeloablative allogene SCT 56% und 62%, nach RIC 49% und 64%. Das Auftreten einer chronischen Graft-versus-Host-Krankheit ging in dieser Studie einher mit einer verbesserten progressionsfreien Überlebensrate als Hinweis auf einen klinisch relevanten Graft-versus-Waldenström-Effekt [37, 38].
Neue Therapieansätze
Einer der erfolgversprechendsten neuen Therapieansätze ist die Blockade der Bruton-Tyrosinkinase (BTK) durch Ibrutinib. In einer amerikanischen Phase-II-Studie mit 63 symptomatischen rezidivierten oder refraktären Patienten mit Morbus Waldenström konnte die tägliche orale Applikation von 420 mg Ibrutinib bei den meisten Patienten ein rasches Ansprechen erzielen: Die mediane Zeit, um zumindest eine „Minor Response“ zu erzielen, betrug nur vier Wochen, die Gesamtansprechrate 90,5%, die „Major Response“-Rate betrug 73,0%. Interessanterweise war das Ansprechen am besten bei Patienten mit der Mutation MYD88(L265P) und Vorliegen eines CXCR4–Wildtyps (100% Gesamtansprechen und 91,2% „Major Response“-Rate), schlechter bei Patienten mit der MYD88(L265P)-Mutation und mutiertem CXCR4 (85,7% Gesamtansprechen und 61,9% „Major Response“-Rate) und am schlechtesten bei Patienten mit unmutiertem MYD88 und CXCR4 (nur 71,4% Gesamtansprechen und 28,6% „Major Response“). Das geschätzte progressionsfreie 2-Jahres-Überleben betrug 69,1%, das Gesamtüberleben zum gleichen Zeitpunkt 95,2% für die Gesamtgruppe. Das Nebenwirkungsprofil bestand zumeist aus Neutropenien (> Grad 2 bei 22% der Patienten) und Thrombozytopenien (bei 14%; [39]).
Eine weitere vielversprechende Substanz ist der PI3Kδ-Inhibitor Idelalisib: In einer Phase-II-Studie mit Patienten, die sowohl gegen Rituximab als auch gegen Alkylanzien bereits refraktär waren, konnte mit der Substanz in der Subgruppe der Waldenström-Patienten eine „Major Response“-Rate von 75% erzielt werden [40]. Allerdings war die Anzahl der behandelten Patienten mit Morbus Waldenström in dieser Studie gering (n = 10), sodass weiterführende Studien Effektivität und Toxizität dieses innovativen Therapieansatzes bei dieser Erkrankung testen müssen.
Behandlungsalgorithmen bei Morbus Waldenström
Basierend auf den oben genannten Behandlungsprinzipien werden derzeit von der Deutschen Gesellschaft für Hämatologie und Onkologie (DGHO; www.mein-onkopedia.de) und der European Society for Medical Oncology (ESMO) folgenden Behandlungsalgorithmen für die Erstlinien – (Abb. 1) und für die Rezidivtherapie (Abb. 2) vorgeschlagen, wobei jedoch individuelle Eigenschaften des Patienten berücksichtigt werden sollten.
Therapiekontrolle und Nachsorge
Unter der Behandlung sind regelmäßige Therapiekontrollen zur Erkennung und Bewertung von Komplikationen und Nebenwirkungen durchzuführen. Hierzu gehören: Anamnese und körperliche Untersuchung, die Anfertigung eines Blut- und Differenzialblutbilds sowie die Bestimmung von LDH, Nieren- und Leberwerten. Eine Therapiebewertung initial pathologischer Befunde (der IgM-Spiegel und der Krankheitsherde, die vor der Behandlung auffällig waren) empfiehlt sich nach der Hälfte der Therapiezyklen sowie nach Abschluss einer zytostatischen Behandlung.
Da der Morbus Waldenström letztlich eine chronische Krankheit ist, sind Rückfälle auch nach Jahren möglich. Besondere Aufmerksamkeit gilt daher der Erkennung von Rezidiven und Sekundär-Neoplasien. In den ersten zwei Jahren nach Erreichen einer Remission sind Verlaufskontrollen als Nachsorge in dreimonatigen Abständen sinnvoll, ab dem dritten Jahr in sechsmonatigen Abständen. Hierzu gehören Anamnese und körperliche Untersuchung, ein Blutbild inklusive Differenzialblutbild, LDH, Nieren- und Leberwerte, IgM-Bestimmung sowie während der ersten zwei Jahre in sechs- bis zwölfmonatigen Intervallen eine Bildgebung bei zuvor pathologischen Befunden (Ultraschall oder radiologische Bildgebung; [16]).
Fazit
Zusammenfassend haben wir immer noch nicht das „Wundermittel“ für die Erkrankung Morbus Waldenström, aber es gibt kontinuierlich Fortschritte bei der Behandlung. Diese Fortschritte basieren auf kontrollierten klinischen Studien, sodass auch weiterhin versucht werden sollte, Patienten – wann immer möglich – in solche klinischen Studien einzuschließen. Dies wird schließlich den Weg zu Behandlungskonzepten bahnen, mit denen sich eine Langzeitkontrolle der Erkrankung erreichen lässt, ohne die Lebensqualität der meist älteren Patienten negativ zu beeinflussen.
Literatur
1. Groves FD et al. Waldenstrom's macroglobulinemia: Incidence patterns in the United States, 1988-1994. Cancer 1998; 82: 1078-81.
2. Waldenström J. Incipient myelomatosis or essential hyperglobulinemia with fibrinogenopenia − a new syndrome? Acta Med Scand 1944; 117: 216-47.
3. Treon SP et al. Characterization of familial Waldenstrom's macroglobulinemia. Ann Oncol 2006; 17: 488-94.
4. Kristinsson SY et al. Risk of lymphoproliferative disorders among first-degree relatives of lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia patients: A population-based study in Sweden. Blood 2008; 112: 3052-6.
5. Treon SP et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med 2012; 367: 826-33.
6. Yang G et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenstrom macroglobulinemia. Blood 2013; 122: 1222-32.
7. Treon SP et al. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood 2014; 123: 2791-6.
8. Cao Y et al. CXCR4 WHIM-like frameshift and nonsense mutations promote ibrutinib resistance but do not supplant MYD88(L265P)-directed survival signalling in Waldenstrom macroglobulinaemia cells. Br J Haematol 2015; 168: 701-7.
9. Buske C,Leblond V. How to manage Waldenstrom's macroglobulinemia. Leukemia 2013; 27: 762-72.
10. Swerdlow SH et al. World Health Organization Classification of Tumors of Haematopoietic and Lymphoid Tissues. IARC Press 2008, Lyon.
11. Owen RG. Developing diagnostic criteria in Waldenstrom's macroglobulinemia. Semin Oncol 2003; 30: 196-200.
12. San Miguel JF et al. Immunophenotypic analysis of Waldenstrom's macroglobulinemia. Semin Oncol 2003; 30: 187-95.
13. Morel P et al. International prognostic scoring system for Waldenstrom macroglobulinemia. Blood 2009; 113: 4163-70.
14. Owen RG et al. Response assessment in Waldenstrom macroglobulinaemia: Update from the VIth International Workshop. Br J Haematol 2013; 160: 171-6.
15. Dimopoulos MA et al. Treatment recommendations for patients with Waldenstrom macroglobulinemia (WM) and related disorders: IWWM-7 consensus. Blood 2014; 124: 1404-11.
16. Buske C et al. Waldenstrom's macroglobulinaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2013; 24 (Suppl 6): vi155-9.
17. Gertz MA et al. Amyloidosis and Waldenstrom's macroglobulinemia. Hematology 2004 (Am Soc Hematol Educ Program): p. 257-82.
18. Ghobrial IM et al. Initial immunoglobulin M 'flare' after rituximab therapy in patients diagnosed with Waldenstrom macroglobulinemia: An Eastern Cooperative Oncology Group Study. Cancer 2004; 101: 2593-8.
19. Treon SP et al. Maintenance rituximab is associated with improved clinical outcome in rituximab naive patients with Waldenstrom Macroglobulinaemia who respond to a rituximab-containing regimen. Br J Haematol 2011; 154: 357-62.
20. Buske C et al. The addition of rituximab to front-line therapy with CHOP (R-CHOP) results in a higher response rate and longer time to treatment failure in patients with lymphoplasmacytic lymphoma: Results of a randomized trial of the German Low-Grade Lymphoma Study Group (GLSG). Leukemia 2009; 23: 153-61.
21. Abonour R et al. Phase II pilot study of rituximab + CHOP in patients with newly diagnosed WM, an Eastern Cooperative Oncology Group trial (study E1A02). Blood 2007; 118(11): 1058A (ASH 2007, Abstract #3616).
22. Rummel MJ et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: An open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013; 381: 1203-10.
23. Leblond V, Choquet S. Fludarabine in Waldenstrom's macroglobulinemia. Semin Oncol 2003; 30: 239-42.
24. Leblond V et al. Multicenter, randomized comparative trial of fludarabine and the combination of cyclophosphamide-doxorubicin-prednisone in 92 patients with Waldenstrom macroglobulinemia in first relapse or with primary refractory disease. Blood 2001; 98: 2640-4.
25. Dhodapkar MV et al. Long-term survival in Waldenstrom macroglobulinemia: 10-year follow-up of Southwest Oncology Group-directed intergroup trial S9003. Blood 2009; 113: 793-6.
26. Laszlo D et al. Rituximab and subcutaneous 2-chloro-2'-deoxyadenosine combination treatment for patients with Waldenstrom macroglobulinemia: Clinical and biologic results of a phase II multicenter study. J Clin Oncol 2010; 28: 2233-8.
27. Treon SP et al. Long-term outcomes to fludarabine and rituximab in Waldenstrom macroglobulinemia. Blood 2009; 113: 3673-8.
28. Tedeschi A et al. Fludarabine plus cyclophosphamide and rituximab in Waldenstrom macroglobulinemia: An effective but myelosuppressive regimen to be offered to patients with advanced disease. Cancer 2012; 118: 434-43.
29. Ghobrial IM et al. Phase II trial of weekly bortezomib in combination with rituximab in relapsed or relapsed and refractory Waldenstrom macroglobulinemia. J Clin Oncol 2010; 28: 1422-8.
30. Dimopoulos MA et al. Primary treatment of Waldenstrom macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide. J Clin Oncol 2007; 25: 3344-9.
31. Dimopoulos MA et al. Update on treatment recommendations from the Fourth International Workshop on Waldenstrom's Macroglobulinemia. J Clin Oncol 2009; 27: 120-6.
32. Ansell SM et al. Diagnosis and management of Waldenstrom macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines. Mayo Clin Proc 2010; 85: 824-33.
33. Kahl BS et al. Bendamustine is effective therapy in patients with rituximab-refractory, indolent B-cell non-Hodgkin lymphoma: Results from a Multicenter Study. Cancer 2010; 116: 106-14.
34. Gertz MA et al. Stem cell transplant for Waldenstrom macroglobulinemia: An underutilized technique. Bone Marrow Transplant 2012; 47: 1147-53.
35. Kyriakou C et al. High-dose therapy and autologous stem-cell transplantation in Waldenstrom macroglobulinemia: The Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2010; 28: 2227-32.
36. Kyriakou C et al. Allogeneic stem-cell transplantation in patients with Waldenstrom macroglobulinemia: Report from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clinical Oncol 2010; 28: 4926-34.
37. Anagnostopoulos A et al. Autologous or allogeneic stem cell transplantation in patients with Waldenstrom's macroglobulinemia. Biol Blood Marrow Transplant 2006; 12: 845-54.
38. Garnier A et al. Allogeneic hematopoietic stem cell transplantation allows long-term complete remission and curability in high-risk Waldenstrom's macroglobulinemia. Results of a retrospective analysis of the Societe Francaise de Greffe de Moelle et de Therapie Cellulaire. Haematologica 2010; 95: 950-5.
39. Treon SP et al. Ibrutinib in previously treated Waldenstrom's macroglobulinemia. N Engl J Med 2015; 372: 1430-40.
40. Gopal AK et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med 2014; 370: 1008-18.

Dr. med. Alexander Grunenberg1
Prof. Dr. med. Christian Buske1,2,3
1 Klinik für Innere Medizin III
Universitätsklinikum Ulm
2 Comprehensive Cancer Center Ulm
3 Institut für Experimentelle Tumorforschung Universitätsklinikum Ulm
Korrespondierender Autor:
Prof. Dr. med. Christian Buske
Koordinator des Europäischen Konsortiums für den Morbus Waldenström (ECWM)
Comprehensive Cancer Center Ulm
Institut für Experimentelle Tumorforschung
Universitätsklinikum Ulm
Albert-Einstein-Allee 11, 89081 Ulm
+49 731 50065-800
+49 731 50065-822
christian.buske[at]uni-ulm[dot]de