Praktische Herausforderungen beim maschinellen Lernen: Auf die Datenaufbereitung kommt es an
DOI: https://doi.org/10.47184/td.2024.01.07In der Laboratoriumsmedizin kann maschinelles Lernen eingesetzt werden, um in den reichlich vorhandenen Daten verborgene Strukturen und Zusammenhänge zu entdecken oder die Diagnosefindung zu unterstützen. Am Beispiel eines Datensatzes aus der Onkologie werden die einzelnen Prozessschritte von den Rohdaten bis zum fertigen Ergebnis demonstriert. Dabei verursacht die Datenaufbereitung den höchsten Aufwand.
Schlüsselwörter: Maschinelles Lernen, Datenaufbereitung, Dimensionsreduktion, Entscheidungsunterstützung
Maschinelles Lernen (ML) ist ein hochaktuelles Teilgebiet der Künstlichen Intelligenz (KI), das in der Laboratoriumsmedizin zunehmend an Bedeutung gewinnt [1]. Im Gegensatz zu traditionellen prozeduralen Computerprogrammen, bei denen der Weg der Problemlösung vom Menschen vorgegeben wird, sollen ML-Programme diesen Weg selbstständig finden, indem sie „aus den Daten lernen“.
Eine typische Aufgabe unseres Fachs besteht beispielsweise darin, aus einem Muster von Laborwerten auf die zugrundeliegende Krankheit zu schließen. Den Lösungsweg kann man im Sinne eines diagnostischen Pfades in Form von Wenn-dann-Regeln ausprogrammieren. Alternativ könnte man dem Computer aber auch eine Datentabelle präsentieren (Abb. 1) und ihn anweisen, einen statistisch validen Entscheidungsbaum vorzuschlagen [2].
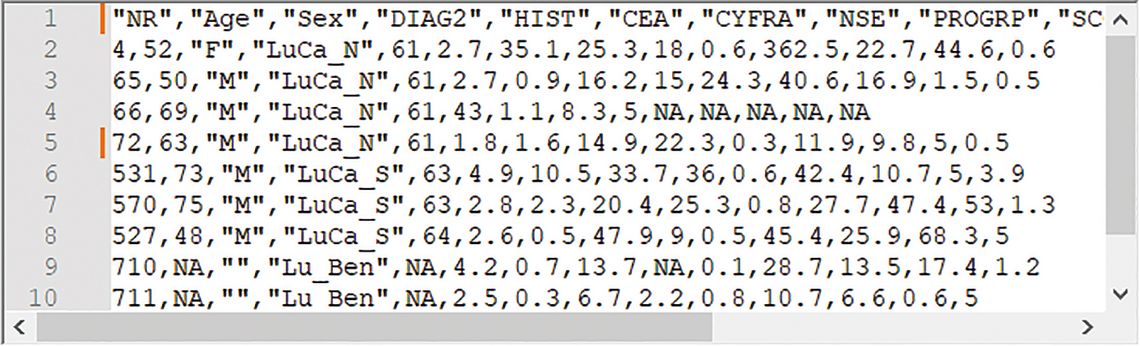
Abb. 1: Ein typischer Originaldatensatz für das maschinelle Lernen. Die Spaltennamen in der ersten Zeile repräsentieren neben demografischen Angaben die histologisch gestellten Diagnosen sowie eine Reihe von Tumormarkern. Die Zeilen entsprechen den untersuchten Personen.
NA = nicht angegeben. Alle Bilder: Autoren.
Beim überwachten Lernen (Supervised Learning) enthält die Tabelle eine Spalte mit vorab festgelegten Zielwerten. Im vorliegenden Fall sind es Klassen (Diagnosen), die aufgrund der Histologie definiert wurden. Beim unüberwachten Lernen (Unsupervised Learning) erhält der Computer keine Angaben zu etwaigen Zielattributen; er soll vielmehr selbstständig interessante Subgruppen oder auch sinnvolle Assoziationsregeln vorschlagen.
In diesem Beitrag wollen wir beide Formen von maschinellem Lernen erläutern, indem wir einen realen onkologischen Datensatz [3] unüberwacht analysieren lassen und prüfen, inwieweit die gefundenen Kategorien (Cluster) mit den histologisch diagnostizierten Lungentumoren übereinstimmen. Zudem wollen wir automatisch einen Entscheidungsbaum erstellen lassen, der benigne Lungenerkrankungen sowie kleinzellige (SCLC) und nicht-kleinzellige (NSCLC) Lungenkarzinome anhand von Tumormarkern unterscheidet.
Datenaufbereitung
Es liegt auf der Hand, dass die Datenaufbereitung den entscheidenden Schritt beim maschinellen Lernen darstellt, denn die Qualität des Ergebnisses hängt maßgeblich von der Datenqualität ab (Garbage in, Garbage out). So wie die Präanalytik im medizinischen Labor den höchsten Arbeitsaufwand verursacht und die größte Fehlerquelle darstellt, ist die Datenaufbereitung beim maschinellen Lernen der herausforderndste und fehlerträchtigste Schritt. Die eigentlichen Algorithmen der KI können dagegen häufig mit einer einzigen Programmzeile aufgerufen werden, so wie man im automatisierten Labor oftmals nur auf den Startknopf drücken muss, um die Analytik durchzuführen.
Tab. 1 gibt einen Überblick über wichtige Schritte der Datenaufbereitung für das maschinelle Lernen.
Tab. 1: Wichtige Schritte der Datenaufbereitung beim maschinellen Lernen in der Labormedizin
Schritt | Ziel | Herausforderungen |
|---|---|---|
Import und Integration | Daten aus verschiedenen Quellen zusammenführen | z. B. manuelle Eingaben, mangelnde Standardisierung |
Exploration (EDA) | Dateneigenschaften, Muster und Beziehungen verstehen | Abgleich mit den Zielen der Studie, Datenqualität |
Bereinigung | Datenqualität verbessern | Fehlende Werte, Duplikate, Ausreißer, Inkonsistenzen |
Transformation und Skalierung | Daten für die Modellbildung und Auswertung aufbereiten | Unterschiedliche Vorgehensweise je nach Fragestellung |
Dimensionsreduktion | Komplexe Daten visualisieren, Komplexität verringern | Informationsverlust, Auswahl der richtigen Verfahren |
Für eine ausführliche Darstellung der Herausforderungen und Lösungen sei auf die Empfehlungen der IFCC zum maschinellen Lernen in der Labormedizin verwiesen [4].
Die Datenaufbereitung kommt zwar zeitlich vor dem eigentlichen maschinellen Lernen, aber sie muss stets „vom Ende hergedacht werden“. Das heißt: Ohne vorab definiertes Ziel (Business Understanding) kann man keine Aussagen darüber machen, welche Techniken der Datenaufbereitung zum Einsatz kommen sollen. Möchte man beispielsweise nur Muster in unstrukturierten Daten entdecken, so kann man den Originaldatensatz häufig komplett an das ML-Programm übergeben. Soll der Computer hingegen lernen, verschiedene Krankheitsbilder auf der Basis solcher Muster zu unterscheiden, so besteht einer der ersten Schritte darin, die Originaldaten in einen Trainings- und einen Testdatensatz aufzutrennen (Data Splitting), die dann separat nach identischen Prinzipien aufbereitet werden.
In der Regel werden ML-Applikationen aus verschiedenen Datenquellen gespeist, die man zu einer einzigen Datentabelle zusammenfasst (Data Integration). Sie ist nach dem „Tidy Data“-Konzept aufgebaut (tidy = ordentlich): Variablen stehen in Spalten, Fälle in Zeilen, und jede Zelle enthält nur einen Wert. Abb. 1 zeigt so eine Tabelle im maschinenlesbaren csv-Format. Sie enthält durch Kommas getrennt Diagnosen, histologische Codes, Laborwerte und demografische Daten.
Typische Herausforderungen bei diesem Arbeitsschritt sind inhomogene Formate der einzulesenden Quelldateien (xlsx, csv, html usw.), mangelnde Standardisierung der Daten (v. a. bei klinischen Angaben) sowie etwaige manuelle Eingaben, die viele Kontrollen und Korrekturen nach sich ziehen können.
Exploration
Als explorative Datenanalyse (EDA) bezeichnet man die detaillierte Untersuchung und Visualisierung der Daten mit dem Ziel, ein besseres Verständnis ihrer Eigenschaften zu gewinnen. Dazu gehören statistische Kenndaten wie etwa Mittelwerte und Standardabweichungen, Verteilungsanalysen mit Boxplots, Histogrammen und Dichtekurven, Erkennung von Mustern und Zusammenhängen in den Daten u. v. m. Dieser als Data Understanding bezeichnete Prozess schafft die Grundlagen für die Entwicklung von ML-Modellen, die geeignet sind, die vorab definierte Studienfrage zu beantworten.
In Zeile drei der Tabelle geht es darum, die Datenqualität gezielt zu verbessern, damit die ML-Algorithmen reibungslos ablaufen und aussagekräftige Resultate liefern. So können viele Funktionen nicht mit fehlenden Daten umgehen. Dafür gibt es zwei Lösungen:
- Löschen der Zeilen mit fehlenden Werten (Cave: Informationsverlust)
- Einfügen von Werten (Imputation)
Das Einfügen erfordert ein gutes Verständnis des Datensatzes im Kontext der Datenerhebung. So macht es einen Unterschied, ob das Fehlen von Daten auf reinem Zufall beruht (z. B. defektes Analysengerät) oder einen informationshaltigen Grund hat (Kranke gehen aus Angst vor schlechten Nachrichten nicht zum Check-up). Im ersten Fall kann man beispielsweise Mediane einfügen, ohne die Auswertung systematisch zu verzerren, im zweiten Fall sind aufwendige statistische und medizinische Überlegungen erforderlich, auf die hier nicht eingegangen werden kann.
Transformation und Skalierung
Das Ziel der Datentransformation ist es, die Originaldaten in eine Form zu bringen, die für die Verarbeitung durch die Lernalgorithmen optimal ist. So erwartet die Hauptkomponentenanalyse (s. u.) zum Beispiel eine annähernde Normalverteilung. Sind die Daten wie im Fall der Tumormarker rechtsschief verteilt, so empfiehlt sich das Logarithmieren, um die Symmetrie der Dichtekurven zu verbessern.
Die Skalierung dient dazu, die Werte verschiedener Variablen untereinander vergleichbar zu machen. Das häufigste Verfahren ist die z-Score-Skalierung, die angibt, um wie viele Standardabweichungen ein Wert vom Mittelwert des Kollektivs abweicht. In unserem Datensatz liegen die Absolutwerte des Tumormarkers ProGRP zwischen 2 und 109.000 pg/ml, für SCC dagegen zwischen 0,1 und 176 ng/ml. Nach der z-Score-Skalierung der logarithmierten Werte bewegen sie sich auf vergleichbaren Skalen zwischen etwa -2 und +5 (Abb. 2).
Abb. 2: Transformation und Skalierung. Links Absolutwerte, rechts z-Scores der logarithmierten Werte.
Die letzte Zeile von Tab. 1 befasst sich mit „höherdimensionalen Datenräumen“. Das klingt geheimnisvoller als es ist. Einen zweidimensionalen Datenraum kann man sich gut als Punktewolke mit beispielsweise ProGRP auf der x-Achse und SCC auf der y-Achse vorstellen. Möchte man aber alle neun Tumormarker in einem einzigen Diagramm visualisieren, so stoßen nicht nur die Drucktechnik, sondern auch das menschliche Vorstellungsvermögen an ihre Grenzen.
Deshalb wurden zahlreiche Verfahren zur „Dimensionalitätsreduktion“ wie etwa die Hauptkomponentenanalyse (PCA = Principal Component Analysis) entwickelt. Das Ziel ist es einerseits, höherdimensionale Daten zu visualisieren, und andererseits auch die Komplexität solcher Daten für ML-Algorithmen zu verringern. Ohne auf mathematische Details einzugehen, besteht der Trick darin, die Anzahl der Variablen (hier also Tumormarker) dadurch zu reduzieren, dass man mehrere davon mit statistischen Verfahren zu neuen Variablen zusammenfasst. Bei der PCA sind dies die Hauptkomponenten. Andere Verfahren verwenden andere Verdichtungstechniken. Entscheidend ist immer, dass auch bei reduzierter Komplexität möglichst viel relevante Information erhalten bleibt.
Zwei Praxisbeispiele
Zum Schluss wollen wir unseren aufbereiteten Datensatz für unüberwachtes und überwachtes maschinelles Lernen einsetzen. Er enthält dank Imputation von Medianen keine fehlenden Werte mehr und ist sowohl logarithmiert als auch skaliert.
Im ersten Experiment suchen wir mit dem k-means-Algorithmus nach Patientengruppen mit ähnlichen Wertekonstellationen. Abb. 3 zeigt in der zweidimensionalen PCA-Darstellung drei Cluster.
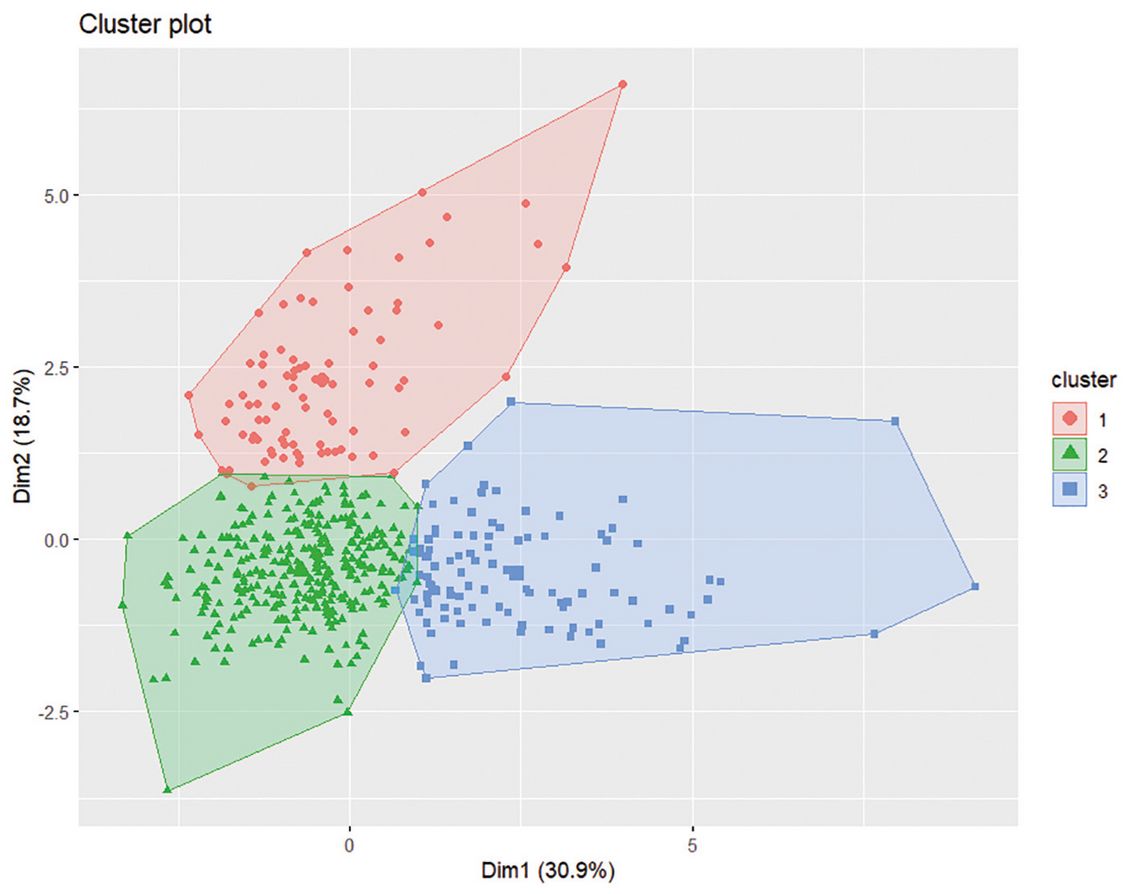
Abb. 3: Unüberwachtes Lernen mit dem k-means-Algorithmus.
Der Vergleich mit den drei Diagnosegruppen ergibt: Der rote Cluster enthält fast ausschließlich kleinzellige Karzinome (SCLC), der blaue fast nur nicht-kleinzellige Karzinome (NSCLC). Der grüne Cluster ist vergleichsweise heterogen: Hier finden sich sämtliche benignen Kontrollen sowie rund zwei Drittel der NSCLC und ein Drittel der SCLC. Dabei handelt es sich vorwiegend um Tumoren mit relativ niedrigen Werten, die von den Kontrollen nur schlecht zu unterscheiden sind.
Im zweiten Experiment bieten wir dem Computer auch die drei Diagnosen an und erhalten mit dem rpart-Algorithmus den in Abb. 4 gezeigten Entscheidungsbaum.
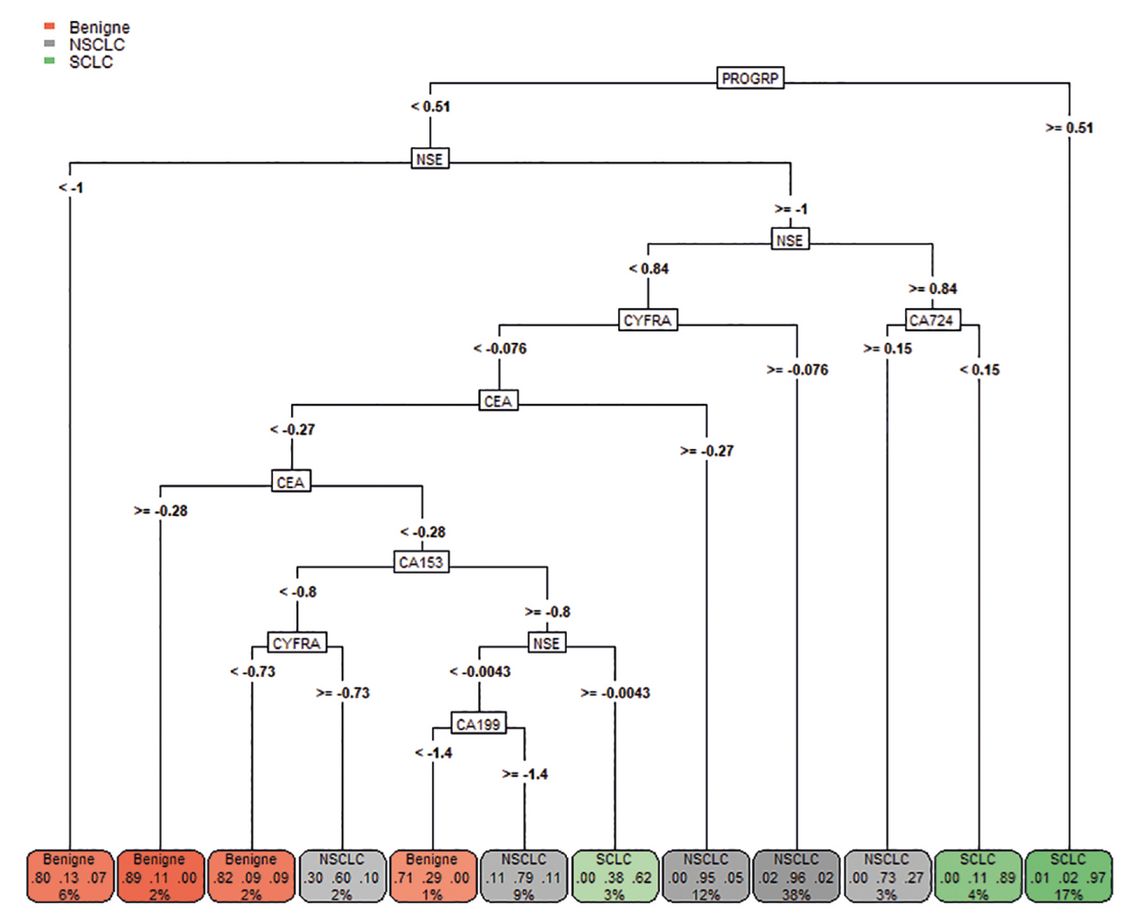
Abb. 4: Überwachtes Lernen mit dem rpart-Algorithmus.
Dieser steht im Einklang mit der Literatur: Erhöhte ProGRP- und NSE-Werte deuten auf SCLC hin, während niedrige Werte auf eine benigne Erkrankung schließen lassen. NSCLC wird durch erhöhte Werte von CEA und CYFRA nahegelegt. In der Leave-one-out-Validierung [2] werden 89 % der Fälle richtig klassifiziert.
