
Ein stiller Held
Das Komplementsystem
Das mehr als 30 verschiedene Proteine umfassende Komplementsystem spielt eine zentrale Rolle in der angeborenen Immunabwehr und ist an zahlreichen Krankheitsbildern ursächlich beteiligt. Es gibt zwar eine Reihe labordiagnostischer Tests, doch die Möglichkeiten sind noch bei Weitem nicht ausgeschöpft.
Schlüsselwörter: Komplementsystem, Infektion, Autoimmunerkrankungen
Wie wichtig die Feuerwehr ist, die man oft das ganze Jahr nicht bemerkt, sieht man erst, wenn es brennt – und ganz Ähnliches gilt für das angeborene Immunsystem: Es steht als universelle Schutztruppe permanent in Bereitschaft und greift ein, wenn der Organismus vor potenziell schädlichen Substanzen, Zellen oder Mikroorganismen geschützt werden muss. Zu diesen „stillen Helden“ zählen neben Endothelzellen, Granulozyten, Makrophagen und NK-Zellen vor allem auch zahlreiche Proteine des Blutes.
Eine ganz wesentliche Rolle in diesem komplexen Konzert spielen die mehr als 30 Plasmaproteine, die man unter dem Oberbegriff Komplementsystem (KS) zusammenfasst. Einige von ihnen fungieren als Proteasen, andere als Rezeptoren, Inhibitoren oder Stabilisatoren. Manche Komponenten haben Lektin-ähnliche Eigenschaften: Sie können an spezifische Kohlenhydratstrukturen von Bakterien und anderen Pathogenen binden und eine Reaktionskaskade auslösen – ähnlich wie dies auch vom Gerinnungssystem her bekannt ist. Das KS kommuniziert in der Tat eng mit den Mechanismen der Hämostase, aber auch mit der Blutdruckkontrolle und vielen anderen Regulationsvorgängen im Körper, und einige seiner Komponenten wie zum Beispiel der C1-Inhibitor (C1-INH) sind in mehreren dieser Systeme aktiv.
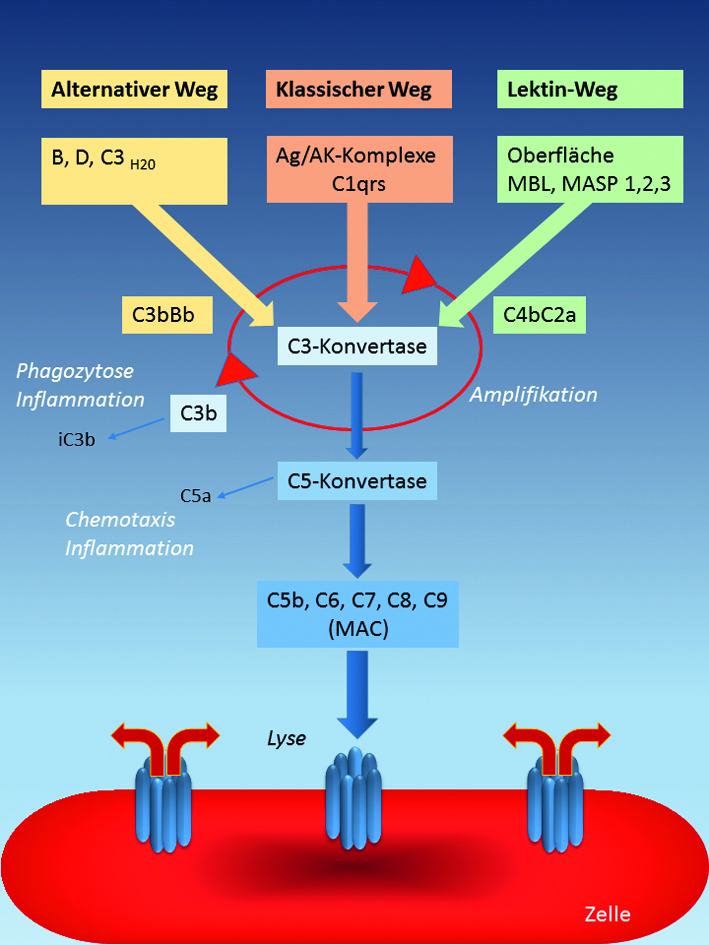
Drei Signalwege
Das KS wird erst bei Bedarf „scharf gestellt“. Seine Aktivierung erfolgt über drei sich gegenseitig ergänzende und verstärkende Wege (Abb. 1), die nach der Aktivierung des Komplementfaktors C3 in eine gemeinsame Endstrecke (C5 bis C9) münden. Nur der sogenannte alternative Weg ist durch spontanen Zerfall von C3 in C3a und C3b permanent auf geringem Niveau aktiv; die beiden anderen – der klassische und der Lektin-Weg – werden durch Antigen-Antikörper-Komplexe bzw. fremde Oberflächenstrukturen, beispielsweise von Bakterien, getriggert.Diese drei Reaktionskaskaden generieren eine Fülle von biologisch aktiven Komponenten, die allerdings nicht nur für die erwünschten Abwehrleistungen, sondern auch für Krankheitssymptome verantwortlich sein können. Dazu gehören beispielsweise die Anaphylatoxine C3a, C4a, und C5a, die die Gefäßpermeabilität erhöhen, glatte Muskeln kontrahieren und aus Mastzellen Histamin freisetzen. Daraus können dann schwere Entzündungssymptome bis hin zum anaphylaktischen Schock resultieren.
Wirkungen und Nebenwirkungen
Eine Hauptaufgabe des KS liegt in der sogenannten Opsonierung von Krankheitserregern: Aktivierte Zwischenprodukte der Kaskade (C3b, iC3b, C4b) binden an die Zelloberfläche und markieren die Eindringlinge für den Angriff durch phagozytierende Abwehrzellen (Chemotaxis). Zusätzlich leistet das KS auch einen eigenständigen Beitrag zur Abwehr, denn seine Endstrecke mündet in der Bildung des terminalen Membranangriffskomplexes (MAC) aus den Komponenten C5b bis C9. Dieser bohrt Löcher in die Zellmembran und induziert so die Lyse der Zellen und damit deren Untergang.Ist dieser Prozess erst einmal in vollem Gange, so kann es aufgrund des relativ unspezifischen Angriffs durchaus zu „Kollateralschäden“ kommen. Sie resultieren vor allem aus der Freisetzung von Inhaltsstoffen aus lysierten Zellen. Beispielsweise reagiert freies Hämoglobin aus Erythrozyten mit dem hormonähnlich wirksamen Stickstoffmonoxid (NO) und induziert damit eine Vasokonstriktion, was Störungen der Organdurchblutung zur Folge haben kann. Freigesetzte DNA und Histone aktivieren das Gerinnungssystem und können so Thrombosen auslösen. Ganz generell interagieren Komplement- und Gerinnungssystem im Sinne positiv rückgekoppelter Regelkreise: MASP-1 und MASP-2 des Lektinwegs aktivieren Komponenten der Hämostase und umgekehrt können einige Gerinnungsfaktoren Komplementkomponenten aktivieren. Solche Verstärkermechanismen sind wahrscheinlich auch ursächlich an der massiven Entzündung bei Sepsis beteiligt.
Im Normalfall unterliegt das KS allerdings einer engen Kontrolle, um eine permanente oder überschießende Aktivierung zu vermeiden: Seine Komponenten liegen im Plasma als inaktive Zymogene vor, spontan aktivierte Moleküle, die nicht an Membranen gebunden sind, werden rasch hydrolysiert, der klassische Weg wird durch den C1-Inhibitor gehemmt, und die Faktoren H und I sowie Faktor-H-ähnliche Proteine (CFHR 1 bis 5) schützen vor unspezifischer Komplementaktivierung. Bei der Regulation der Anaphylatoxine C3a und C5a ist der Thrombin-abhängige Fibrinolyseinhibitor (TAFI) von entscheidender Bedeutung.
Komplementmangelzustände
Mängel sind für nahezu alle Komplementproteine beschrieben (Tabelle 1). Hereditäre Komplementdefekte machen bis zu 6% aller primären Immunmangelzustände aus und werden überwiegend autosomal rezessiv vererbt. Deshalb sind heterozygote Träger klinisch meist unauffällig. Besonders häufig findet man mutationsbedingte Mängel des MBL (Mannose- bzw. Mannan-bindendes Lektin im alternativen Weg) mit einer Prävalenz von bis zu 10% in Europa.Erworbene Mängel entstehen in der Regel durch Überaktivierung bei Entzündungen mit nachfolgendem Verbrauch, seltener durch verminderte Synthese bei Lebererkrankungen. Verlaufsbeobachtungen von C3c oder Properdin empfehlen sich zur Identifizierung von Risikopatienten, zum Beispiel bei Infektionen mit Neisserien, nach Polytrauma und Verbrennungen, bei extrakorporaler oder immunsuppressiver Therapie sowie nach Transplantationen.Klinisch äußert sich ein Mangel an Komplementkomponenten vor allem in erhöhter Infektanfälligkeit im Kindesalter (Atemwege, Mittelohr, Darm) sowie einem vermehrten Auftreten von Lupus erythematodes und anderen Autoimmunerkrankungen.
Überschießende Aktivierung
Zu einer ständigen Überaktivierung des KS kommt es beim angeborene Mangel des C1-Inhibitors. Die betroffenen Patienten leiden unter zum Teil lebensbedrohlichen Angioödemen (Schwellungen) im Magen-Darmtrakt und Kehlkopfbereich mit Durchfällen bzw. akuten Erstickungszuständen. Diese werden durch medikamentöse Trigger oder auch spontan ausgelöst, und zwar als Folge einer überschießenden Aktivierung des Kallikrein-Kinin-Systems mit Freisetzung von Bradykinin. Zur Behandlung werden im akuten Fall C1-INH-Konzentrate, der Bradykininrezeptorantagonist Icatibant oder auch anabole Steroide wie Danazol eingesetzt. Ebenfalls lebensbedrohlich können hämolytische Krisen verlaufen, zum Beispiel beim hämolytisch-urämischen Syndrom (HUS). Neben infektiösen Ursachen kommt auch ein hereditärer Mangel an funktionsfähigem Faktor H (z. B. Y402H-Polymorphismus) sowie selten eine erworbene Spontanmutation des PIG-A-Gens (paroxysmale nächtliche Hämoglobinurie) infrage. Zu den Therapieoptionen gehören die Plasmapherese zur Entfernung von MAC, die Zufuhr von Faktor H mittels Frischplasma und die Hemmung von aktiviertem C5 durch einen monoklonalen Antikörper (Eculizumab).

Dr. Hans-Jürgen Kolde
Mitglied der Redaktion