Neuigkeiten zu akuten Leukämien
ASCO (Chicago), EHA (Madrid) 2017
Akute Leukämien waren vor allem beim diesjährigen Kongress der European Hematology Association (EHA) in Madrid wieder eine wichtige Indikationsgruppe mit mehreren Sitzungen; einige Resultate wurden auch bei der Jahrestagung der American Society of Clinical Oncology (ASCO) in Chicago vorgestellt. Bei der akuten myeloischen Leukämie (AML) standen dabei vor allem molekulare Subgruppen und die Versuche im Vordergrund, diese mit spezifischen Inhibitoren zu behandeln, während bei der akuten lymphatischen Leukämie (ALL) derzeit Immuntherapien und darunter vor allem T-Zellen mit chimärem Antigenrezeptor (CAR-T-Zellen) an vorderster Front stehen.
AML
Rezidivierte oder refraktäre FLT3-mutierte AML
Die akute myeloische Leukämie (AML) zerfällt zunehmend in immer mehr (und größtenteils immer kleinere) molekular definierte Subgruppen mit ganz unterschiedlicher Prognose. Versuche, die Charakterisierung molekulargenetischer Defekte in neue, zielgerichtete Therapien umzumünzen, scheinen in einigen Bereichen langsam Früchte zu tragen. Dazu zählt die Subgruppe der AML-Erkrankungen mit Mutationen des FLT3-Rezeptors, vor allem mit den ITD-Mutationen (Internal Tandem Duplications, ca. 30% aller Patienten mit AML): Ein relativ unspezifischer FLT3-Inhibitor, Midostaurin, konnte in der Phase-III-Studie RATIFY, als Konsolidierungstherapie im Anschluss an eine klassische
7 + 3-Induktionstherapie gegeben, das Ansprechen von Patienten mit mutiertem FLT3 vertiefen und – das war der primäre Endpunkt der Studie – die Überlebenschancen der Patienten verbessern [1]. In den USA ist Midostaurin für diese Indikation bereits zugelassen, in Europa steht die Zulassung kurz bevor.
Eine weitere Substanz, Gilteritinib (ASP2215), das im Gegensatz zu Midostaurin hochselektiv nur FLT3 und AXL hemmt, hat in der Phase-I/II-Studie CHRYSALIS in Dosierungen von 80 mg/d und darüber anti-leukämische Aktivität in der rezidivierten/refraktären Situation bewiesen. In einer exploratorischen Analyse, die Jessica Altman, Chicago, beim ASCO- sowie beim EHA-Kongress vorstellte [2, 3], wurde der Einfluss der minimalen Resterkrankung (MRD) auf die Prognose untersucht. Die MRD-Bestimmung erfolgte mittels Next Generation Sequencing (NGS) aus Knochenmark vor Beginn und mindestens einmal im Verlauf der Therapie bei den Patienten mit FLT3-Mutationen, die mit 120 oder 200 mg/d Gilteritinib behandelt wurden. Bei diesen Dosierungen hatten sich eine konsistente, kontinuierliche FLT3-Inhibition und hohe klinische Ansprechraten gezeigt.
Wurden die Patienten anhand des molekularen Ansprechens in Kategorien eingeteilt, so zeigte sich, dass die mediane Überlebensdauer umso länger war, je tiefer die molekulare Remission: Patienten, die unter der Gilteritinib-Therapie MRD-negativ wurden, in deren Knochenmark sich also weniger als eine FLT3-ITD-mutierte Zelle pro 10.000 Leukozyten mit normalem FLT3 fand, überlebten median 59,6 Monate lang, während diejenigen, die diese Remissionstiefe nicht erreichten, es nur auf median
30,4 Monate brachten (p = 0,002).
Damit, so Altman, konnte zum ersten Mal nachgewiesen werden, dass die AML-Therapie mit einem FLT3-Inhibitor zu einer tiefen molekularen Remission führen kann. Der Zusammenhang lässt vermuten, dass – ähnlich wie das für andere hämatologische Erkrankungen auch schon gezeigt werden konnte – das molekulare Ansprechen mit der Prognose und mit einem dauerhaften klinischen Nutzen der Gilteritinib-Behandlung korreliert.
Besseres Ansprechen und Überleben auch mit Quizartinib
Patienten mit einer ITD-Mutation des FLT3-Gens/-Proteins haben ein hohes Rezidivrisiko und eine schlechte Prognose: In einer früheren Studie lag ihr medianes Überleben nach einer zweiten Salvagetherapie bei lediglich 1,5 Monaten [4]. Der potente und hochselektive FLT3-Inhibitor Quizartinib konnte in einer einarmigen Phase-II-Studie bei Patienten mit FLT3-ITD-mutierter AML, die nach der Zweitlinientherapie rezidiviert oder refraktär waren, immerhin 23 Wochen erreichen, bei einer Remissionsrate von 46% [5]. Da Patienten mit einem so hohen Risiko von einer allogenen Stammzelltransplantation profitieren, wurden im Kollektiv der genannten Phase-II-Studie 58 Patienten selektiert, die eine intensive chemotherapeutische Vorbehandlung erfahren hatten und vor Eintritt in die Quizartinib-Studie unter einer Salvagetherapie rezidiviert (n = 53) oder refraktär gewesen waren (n = 5; [6]). Sie wurden mit 118 Patienten verglichen, die nach den gleichen Kriterien aus der Datenbank des National Cancer Research Institute und des Medical Research Council von Großbritannien 118 Patienten ausgewählt wurden und lediglich konventionelle Chemotherapien erhalten hatten, bevor bei ihnen über die Möglichkeit einer Transplantation entschieden worden war.
Die mit Quizartinib behandelten Patienten, so Robert Hills, Cardiff, beim EHA-Kongress, zeigten gegenüber den historischen Kontrollen eine signifikant höhere Rate an Remissionen, die vor allem in Komplettremissionen ohne vollständige hämatologische Wiederherstellung bestanden (40% vs. 3%; adjustierte Odds Ratio 0,05; p < 0,0001) und ein deutlich verlängertes medianes Überleben (140 vs. 54 Tage; adjustierte Hazard Ratio 0,38; p < 0,0001). 40% der mit Quizartinib behandelten Patienten gegenüber nur 8% der Kontrollen konnten eine allogene Transplantation erhalten, die in einer Landmark-Analyse die 18-Monats-Überlebensrate vervierfachen konnte (von 7% auf 29%; adjustierte Hazard Ratio 0,36; p = 0,0005).
Da der Vergleich mit historischen Kontrollkollektiven statistisch problematisch ist, wurden Sensitivitätsanalysen durchgeführt, die den Zusammenhang zwischen längerem Überleben und allogener Stammzelltransplantation erhärteten. Eine künftige Rolle von Quizartinib könnten also darin bestehen, so Hills, rezidivierten oder refraktären Patienten mit FLT3-ITD-mutierter AML als Bridging-Option zur allogenen Transplantation zu dienen.
Sorafenib zur Erhaltung nach allogener Transplantation?
Auch nach einer allogenen Transplantation haben AML-Patienten mit FLT3-ITD-Mutation ein höheres Rezidivrisiko und im Rezidiv eine schlechtere Prognose als andere Patienten. Der Multi-Kinaseinhibitor Sorafenib konnte in der Phase-III-Studie SORAML als Zugabe zur konventionellen Chemotherapie bei unter 60-jährigen Patienten ereignis- und progressionsfreies, nicht aber das Gesamtüberleben verlängern. Sorafenib hemmt unter anderem auch die Kinase des FLT3-Rezeptors, und es gibt Hinweise darauf, dass es bei Patienten mit FLT3-Mutationen als Erhaltungstherapie nach einer Transplantation von Nutzen sein könnte. Kollegen am M. D. Anderson Cancer Center in Houston fanden unter den
214 Patienten mit FLT3-ITD-Mutation, die an ihrem Zentrum zwischen 2010 und 2016 transplantiert worden waren,
13 Fälle, in denen eine solche Erhaltung gegeben worden war und verglichen sie mit 26 Patienten, die keine Erhaltungstherapie bekommen hatten, aber ansonsten vergleichbar waren [7].
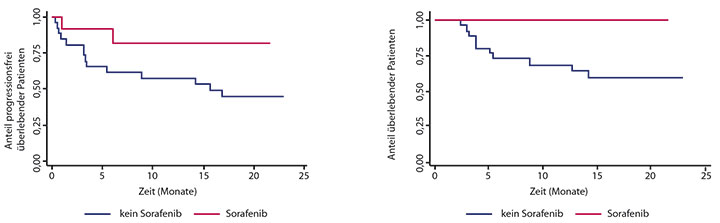
In einer Landmark-Analyse 24 Monate nach Transplantation waren laut Betul Oran, Houston, 82% der mit Sorafenib behandelten, aber nur 45% der Kontroll-Patienten noch progressionsfrei am Leben (Abb. 1 links), die Gesamtüberlebensraten lagen bei 100% bzw. lediglich 60% (Abb. 1 rechts). Von den zwei Patienten, die unter oder nach der Sorafenib-Erhaltungstherapie rezidiviert waren, wies einer im Rezidiv eine neue TP53-Mutation auf, der andere hatte den Inhibitor weniger als 30 Tage lang erhalten. Sorafenib war insgesamt gut verträglich: Fünf Patienten konnten 400 mg/d erhalten, zwei sogar höhere Dosierungen, während sechs sich auf 300 mg/d oder weniger beschränken mussten.
Alles in allem, so Oran, scheint die Erhaltungstherapie mit dem Multi-Kinaseinhibitor Sorafenib sicher und geeignet zu sein, bei diesen Hochrisiko-Patienten eine Remission nach allogener Stammzelltransplantation deutlich zu verlängern.
Neue Option für die IDH2-mutierte AML
Etwas weniger häufig als FLT3-Mutationen, nämlich in 8–15% der Fälle, treten bei der AML Mutationen im Gen für die Isocitrat-Dehydrogenase 2 (IDH2) auf. Das mutierte IDH2-Enzym synthetisiert 2-Hydroxyglutarat, das als Onkometabolit die Hypermethylierung von DNA und Histonen bewirkt und dadurch die Differenzierung myeloider Zellen verhindert. Derzeit sind mehrere Inhibitoren von IDH2 in der klinischen Prüfung, darunter der orale, selektiv wirksame Hemmstoff Enasidenib (AG-221). Er wurde in einer Phase-I/II-Studie bei insgesamt
239 Patienten mit IDH2-mutierter AML getestet, wobei in der Dosiseskalations-Phase mit 650 mg/d die maximal tolerierte Dosierung noch nicht erreicht war, so Eytan Stein, New York, beim ASCO- und beim EHA-Kongress [8, 9]. Eine Reduktion der 2-Hydroxyglutarat-Konzentrationen um etwas mehr als 90% wurde unabhängig von der Dosierung erreicht, sodass 100 mg/d als Standarddosis für die weitere Prüfung gewählt wurden.
Die Toxizität war gering mit 12% Grad-3/4-Hyperbilirubinämien und einem IDH-Inhibitor-assoziierten Differenzierungssyndrom (ähnlich dem Retinsäure-Syndrom) vom Grad 3/4 in 7% der Fälle. Die Gesamtansprechrate der Patienten mit rezidivierter oder refraktärer Erkrankung lag bei 40,3% mit 19,3% Komplettremissionen. Die mediane Überlebensdauer betrug für alle Patienten 9,3 Monate, für diejenigen mit kompletter Remission war sie mit 19,7 Monaten doppelt so lang. Ungefähr die Hälfte der Patienten hatte vorher mindestens zwei Therapien ihrer AML bekommen; für diese lag die mediane Überlebenszeit mit 8,0 Monaten nicht viel niedriger als in der Gesamtkohorte.
Mit seiner guten Verträglichkeit, der Induktion von Komplettremissionen und einer medianen Überlebensdauer von insgesamt mehr als neun Monaten stellt Enasidenib einen vielversprechenden Wirkstoffkandidaten für diese nicht ganz seltene Form der AML dar, so Stein. Offenbar besteht der Mechanismus, der der klinischen Wirksamkeit zugrunde liegt, weniger in einem zytotoxischen als vielmehr in einem differenzierenden Effekt auf die Myeloblasten.
Immuntherapien bei der AML
Immuncheckpoint-Inhibitoren haben in den vergangenen Jahren in nahezu der gesamten Onkologie eine erstaunliche Karriere gemacht. Da sich PD-1-positive zytotoxische T-Lymphozyten vermehrt im Knochenmark von Patienten mit AML finden [10] und Azacitidin die Expression dieses Checkpoint-Moleküls verstärkt und dadurch Resistenzen gegen die hypomethylierende Substanz ausgelöst werden können [11], wurde in einer Phase-Ib/II-Studie bei Patienten mit rezidivierter AML eine Kombination aus dem PD-1-Inhibitor Nivolumab und Azacitidin untersucht. Das Ergebnis wurde mit historischen Kontrollen verglichen, die nur Azacitidin erhalten hatten, so Naval Daver, Houston [12].
Von 70 Patienten sprachen 15 (22%) mit einer kompletten Remission, sieben (10%) mit einer hämatologischen Verbesserung und 17 (24%) mit einer Reduktion der Blastenzahl um mindestens 50% an. Das Gesamtüberleben war vorteilhaft gegenüber historischen Kontrollen, die Azacitidin-basierte Salvageprotokolle ohne Checkpoint-Inhibitor erhalten hatten; signifikant war der Vorteil allerdings nur (p = 0,00992), wenn es sich um die erste Salvagetherapie gehandelt hatte. Immunvermittelte Toxizitäten wurden beobachtet, konnten aber mit den inzwischen etablierten Maßnahmen problemlos kontrolliert werden.
Interessanterweise zeigten sich bei den Patienten, die später auf die Therapie ansprachen, zu Beginn höhere Konzentrationen an CD3- und CD8-positiven T-Zellen und niedrigere Titer an regulatorischen T-Zellen. Möglicherweise zeichnet sich damit ein Biomarker zur Selektion von Patienten ab, die bessere Aussichten auf einen Therapieerfolg haben. Ebenfalls interessant: Bei Respondern und Non-Respondern fand sich eine erhöhte Expression des CTLA-4-Checkpoint-Moleküls auf den CD8-positiven Zellen. Das, so Daver, könnte darauf hindeuten, dass möglicherweise eine Kombination aus PD-1- und CTLA-4-Inhibitor noch wirksamer sein könnte. Eine Studie mit der Kombination aus Azacitidin, Nivolumab und Ipilimumab ist ebenso geplant [13] wie eine andere, in der über 65-jährige Patienten mit neu diagnostizierter AML Azacitidin und Nivolumab als Erstlinientherapie erhalten sollen [14].
Venetoclax bei neu diagnostizierter AML
Zur neu diagnostizierten AML gab es in Madrid zwei Studien, in denen der BCL-2-Inhibitor Venetoclax in Regimes integriert wurde, die auch ältere Patienten tolerieren, die für eine Standard-Induktionstherapie nicht mehr geeignet sind.
In eine US-amerikanisch-französisch-australische Phase-Ib-Studie wurden bisher 100 solcher älteren Patienten eingeschlossen, die für eine Standard-Induktionstherapie nicht geeignet waren [15]. Sie erhielten entweder Decitabin oder Azacitidin in der jeweiligen Standarddosierung und zusätzlich Venetoclax, wobei das Einschleichen hier im Gegensatz zum Vorgehen bei der chronischen lymphatischen Leukämie stark beschleunigt wurde, so Keith Pratz, Baltimore: Die Dosierung begann bei 100 mg/d und wurde im 1-Tages-Takt über 200 auf 400 mg/d und in einer Kohorte sogar auf 800 mg/d erhöht, die allerdings nur intermittierend gegeben wurden.
Die Ansprechraten waren mit 76% (Decitabin) und 68% (Azacitidin) in den Venetoclax-400-mg-Gruppen höher als unter 800 mg/d (71% bzw. 60%). Nach median 5,4 Monaten sind 16 der 100 Patienten verstorben.
Das Sicherheitsprofil war ausgezeichnet mit überwiegend hämatologischen Nebenwirkungen. Ein Tumorlyse-Syndrom wurde nicht beobachtet, sodass nun eine Phase-III-Studie mit Venetoclax und Azacitidin geplant ist [16].
In einer weiteren Phase-I/II-Studie wurde die bei älteren Patienten gebräuchliche Therapie mit niedrig-dosiertem Cytarabin mit dem BCL-2-Inhibitor kombiniert, der hier in der Phase I mit bis zu 800 mg/d gegeben wurde; als Dosis für die Phase II wurden 600 mg/d gewählt [17]. Beide Therapien kommen für sich genommen historisch auf Komplettremissionsraten von maximal 19%, sodass die Rate von 54% unter der Kombination in der Population von 61 Patienten sich eindrucksvoll ausnimmt, so Andrew Wei, Melbourne. Die Aktivität des Regimes erweist sich als weitgehend unabhängig von Alter, Krankheitstyp (auch sekundäre AML), zytogenetischem Risiko oder vorheriger Behandlung mit hypomethylierenden Substanzen. Von den 61 bisher auswertbaren Patienten erzielten 37 eine komplette oder partielle Remission, und ihre Überlebenskurve (mit allerdings noch vielen zensierten Patienten) hebt sich eindrucksvoll von der der 24 Non-Responder ab (Abb. 2).
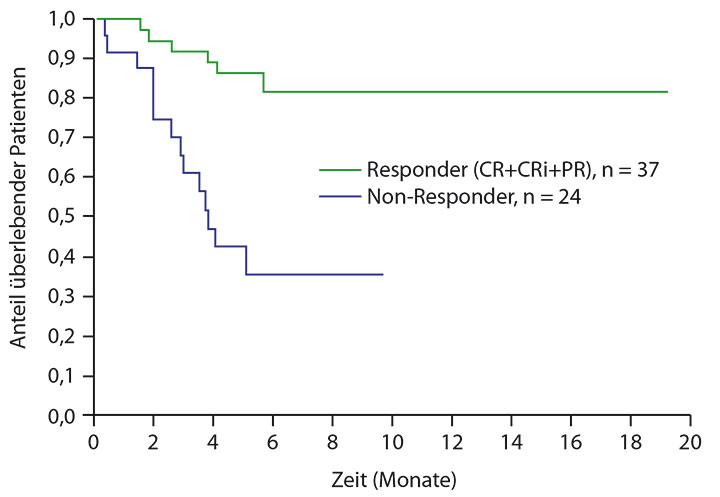
Auch diese Kombination stellt sich sehr vielversprechend dar, so Wei – daher hat eine Phase-III-Studie dazu bereits begonnen [18].
Wie wichtig ist der MRD-Status bei neu diagnostizierter AML?
Eine risikoadaptierte Therapie bewährt sich in der Onkologie generell, um Patienten mit geringem Rezidivrisiko eine Übertherapie zu ersparen und umgekehrt den Hochrisiko-Patienten nicht die Chancen vorzuenthalten, die eine aggressivere Behandlung bietet. Die Risikoeinschätzung erfolgt bei der AML vor allem über die Zytogenetik; hier gibt es drei Kategorien: Ein niedriges Risiko haben NPM1-positive, FLT3-ITD-negative oder Core-Binding-Factor-(CBF)-positive Patienten ohne c-Kit-Mutation, ein hohes Risiko Patienten mit einem ungünstigen Karyotyp oder FLT3-ITD-Mutation. Erstere Gruppe wurde in der Phase-III-Studie AML1310 der italienischen Studiengruppe GIMEMA nach Induktions- und Konsolidierungstherapie mit einer autologen, letztere mit einer allogenen Stammzelltransplantation behandelt, so Adriano Venditti, Rom [19]. Bei der dritten Gruppe, bestehend aus Patienten mit intermediärem Risiko (intermediärer Karyotyp, FLT3-TKD-positiv oder CBF-positiv mit c-Kit-Mutation), wurde die Entscheidung vom MRD-Status nach der Konsolidierung abhängig gemacht: War er positiv, erhielten sie eine allogene, war er negativ, eine autologe Transplantation.
Das Kriterium scheint zuverlässig zu sein: Bei einer medianen Nachbeobachtungszeit von 27,9 Monaten lagen die Gesamt- und die krankheitsfreien 2-Jahres-Überlebensraten im Gesamtkollektiv von 341 Patienten nach der Konsolidierung bei 55,9% bzw. 54,9%, die kumulative Rezidivrate bei 32,9%. Bei Unterteilung in die genannten Subgruppen ergab sich folgendes Bild: In der Niedrigrisiko-Gruppe lebten nach 24 Monaten noch 74,8% der Patienten, davon 63,8% krankheitsfrei, in der Hochrisiko-Gruppe (die allogen transplantiert worden war) waren es 42,5% bzw. 44,8%. In der Gruppe mit intermediärem Risiko waren die Ergebnisse der Patienten mit MRD-Negativität, die also nur autolog transplantiert worden waren, mit 78,6% bzw. 61,4% genauso gut wie im Niedrigrisiko-Arm, bei denen mit positivem MRD-Status, die eine allogene Transplantation erhalten hatten, fielen sie mit 69,8% bzw. 66,6% jedenfalls deutlich besser aus als im Hochrisiko-Arm.
Eine risikoadaptierte Therapie, die sich bei Patienten mit intermediärem Risiko am MRD-Status nach der Konsolidierung orientiert, ist also auch in einem multizentrischen Setting machbar. Der MRD-Status schien sich hier als Entscheidungskriterium zu bewähren: Bei den positiven Patienten kann die allogene Transplantation die Ergebnisse beinahe an die der Niedrigrisiko-Gruppe anpassen, den MRD-negativen Patienten hingegen kann man diese Behandlung ersparen.
Zu anderen Ergebnissen kommt eine Studie der niederländischen HOVON- und der Schweizerischen SAKK-Gruppen: Die Kollegen um Jurjen Versluis, Rotterdam, werteten retrospektiv die Daten von 547 AML-Patienten aus HOVON- und SAKK-Studien aus, die nach zwei Zyklen einer Erstlinien-Induktionstherapie eine erste komplette Remission erzielt hatten und für die zu diesem Zeitpunkt ein durchflusszytometrisch bestimmter MRD-Status zur Verfügung stand [20]. Er war bei 129 von ihnen (24%) positiv; die Risikoklassifikatoren des European LeukemiaNET waren ansonsten gleichmäßig über MRD-positive und -negative Patienten verteilt. Gut die Hälfte der Patienten hatte unabhängig von den MRD-Daten als Post-Remissions-Therapie eine allogene, 105 hatten eine autologe Stammzelltransplantation und 160 einen dritten Chemotherapiezyklus erhalten.
Gesamt- und krankheitsfreies Überleben nach vier Jahren waren bei den MRD-negativen Patienten erwartungsgemäß signifikant besser als bei den MRD-positiven. Das war vor allem durch eine niedrigere kumulative Rezidivrate im MRD-negativen Arm bedingt (32% vs. 54%; p < 0,001), während bei der nicht rezidivbedingten Mortalität kein Unterschied erkennbar war. Interessanterweise ergab eine multivariate Analyse, dass eine allogene Transplantation im Vergleich zu autologer Transplantation oder Chemotherapie das Rezidivrisiko signifikant reduzierte (Hazard Ratio 0,36; p < 0,001), und zwar gleichermaßen bei den MRD-negativen (HR 0,38; p < 0,001) als auch bei den MRD-positiven Patienten (HR 0,35; p < 0,001; Abb. 3). Auch das rezidivfreie Überleben wurde durch die allogene Transplantation im Vergleich zu den beiden anderen Post-Remissions-Behandlungsmodalitäten signifikant verbessert (HR 0,53; p < 0,001), nicht jedoch das Gesamtüberleben.
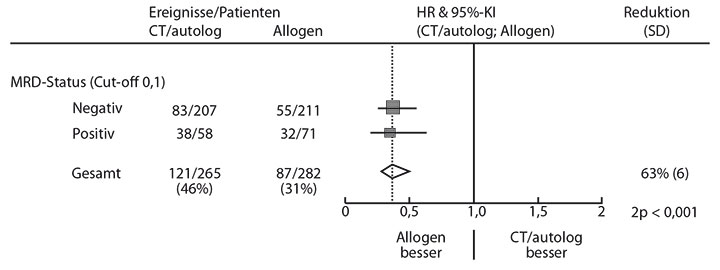
Diese Ergebnisse kratzen etwas an der Vorstellung, dass der MRD-Status das wichtigste Steuerungskriterium bei der Entscheidung für die Konsolidierungstherapie in der ersten Komplettremission sein könnte. Die Autoren interpretieren sie so, dass der Graft-versus-Leukämie-Effekt des allogenen Transplantats auch bei MRD-negativen Patienten noch zum Tragen kommt – was bedeutet, dass auch in dieser Situation noch Leukämiezellen vorhanden sind, die potenziell ein Rezidiv verursachen können. Dies spräche für eine personalisierte Entscheidung für oder gegen die allogene Transplantation, wobei man das Rezidivrisiko, das durch die klassische Risikoklassifikation sowie den MRD-Status bestimmt wird, gegen das Risiko der nicht rezidivbedingten Mortalität abwägen muss.
ALL
Immuntherapien bei der ALL
Die Prognose von Patienten mit rezidivierter oder refraktärer akuter lymphoblastischer Leukämie (ALL) ist ausgesprochen schlecht; die Raten für Komplettremissionen, die alleine eine Heilung ermöglichen würden, werden mit jeder zusätzlichen Behandlungslinie niedriger [21], sodass hier ein dringender Bedarf für neue therapeutische Optionen besteht.
T-Lymphozyten mit chimärem Antigenrezeptor (CAR-T-Zellen) sind in dieser Zeitschrift schon mehrfach ausführlich thematisiert worden. Kurz gesagt haben sie gegenüber der allogenen Stammzelltransplantation den Vorteil, dass sie aus autologen Zellen bestehen und deshalb keine Abstoßungsreaktionen verursachen. Von einer konventionellen autologen Transplantation unterscheidet sie, dass sie mithilfe des rekombinant eingeführten Antigenrezeptors einen starken Graft-versus-Leukämie-Effekt ausüben. Gegenüber einfachen Antikörpern oder Immunkonjugaten haben sie den Vorteil, nach einer einmaligen Gabe potenziell unbegrenzt, aber jedenfalls sehr lange im Körper zu bleiben und aufgrund ihrer Memory-Eigenschaften auch ein späteres Rezidiv erneut bekämpfen zu können.
CAR-T-Zellen werden mittlerweile auch bei Lymphomen, beim multiplen Myelom und bei anderen Leukämien getestet, aber begonnen hat ihre klinische Entwicklung bei der therapierefraktären kindlichen akuten lymphoblastischen B-Zell-Leukämie (B-ALL). Hier laufen daher auch bereits Zulassungsstudien, darunter die Phase-II-Studie ELIANA, in der weltweit bislang 68 pädiatrischen Patienten im medianen Alter von zwölf Jahren mit weit fortgeschrittener B-ALL erfolgreich die an der University of Pennsylvania in Philadelphia entwickelten CAR-T-Zellen mit einem CD19-Antigenrezeptor infundiert wurden [22].
Wie Stephan Grupp, Philadelphia, beim EHA-Kongress berichtete, erzielten 52 der Patienten (83%) innerhalb von drei Monaten nach einer einzigen Infusion der Zellen (zuvor hatten sie eine zytoreduktive Chemotherapie erhalten) eine komplette Remission mit oder ohne vollständige Erholung der hämatologischen Parameter. Alle wiesen auch ein MRD-negatives Knochenmark auf. Sechs Monate nach Einsetzen der Remission waren noch 75% der Patienten rezidivfrei am Leben, die Gesamtüberlebensrate betrug nach sechs Monaten 89% und nach einem Jahr 79%. Sieben Patienten (das sind 13% der Responder) unterzogen sich binnen sechs Monaten einer allogenen Stammzelltransplantation.
Die potenziell bedrohlichste Nebenwirkung bei der Anwendung dieser Therapie ist ein Zytokin-Release-Syndrom, das in dieser Studie auch bei 78% der Patienten auftrat – bei 21% bzw. 27% vom Grad 3 bzw. 4 – aber in keinem Fall zum Tod führte. Es korrelierte mit der Expansion der CAR-T-Zellen im Körper und konnte im Bedarfsfall erfolgreich mit Zytokin-Inhibitoren wie dem Anti-Interleukin-6-Antikörper Tocilizumab behandelt werden. Schwere Nebenwirkungen traten bei 69% der Patienten innerhalb der ersten acht Wochen nach Infusion der Zellen auf, bei 15% neuropsychiatrische Symptome vom Grad 3, aber es kam zu keinem Hirnödem. Elf Patienten sind bisher verstorben, davon sieben an einem Rezidiv oder Progress ihrer ALL. Bei einigen Patienten waren die CD19-CAR-T-Zellen länger als ein Jahr im peripheren Blut nachzuweisen, was jeweils mit einer B-Zell-Aplasie einherging.
Wie bereits in einer Phase-I-Studie und in der beim ASH-Kongress 2016 vorgestellten ersten Interimsanalyse der ELIANA-Studie gezeigt wurde, bestätigen diese Resultate, dass sich mit einer einzigen Infusion dieser Zellen ohne jede weitere anti-leukämische Therapie in verzweifelten Fällen pädiatrischer ALL bemerkenswerte und lang andauernde Remissionen erzielen lassen. Nicht das geringste Verdienst dieser Studie ist der Nachweis, dass die komplexe Logistik auch in einem globalen, multizentrischen Setting funktioniert: Die nativen T-Zellen müssen im gefrorenen Zustand in ein zentrales Labor gesandt, dort mit dem Gen für den Antigenrezeptor versehen, wieder zurückgeschickt und dann dem Patienten infundiert werden. Das alles ist in weniger als drei Wochen machbar, in denen die Leukämie der Patienten gegebenenfalls mit einer Chemotherapie unter Kontrolle gehalten werden muss.
Neurotoxizität nach CAR-T-Zellen
Ein weiteres CD19-CAR-T-Zell-Produkt wurde am Memorial Sloan Kettering Cancer Center in New York entwickelt und in einer Phase-I-Studie an
51 erwachsenen Patienten mit rezidivierter B-ALL getestet. Die Komplettremissionsrate nach vier Wochen hatte 77% für diejenigen betragen, die vor Infusion der Zellen noch mindestens 5% Blasten im Knochenmark aufgewiesen hatten; bei denen mit geringerer Krankheitslast (als “minimale Resterkrankung” bezeichnet) lag sie bei 95%, so Eric Smith, New York [23]. War dieser Unterschied noch nicht statistisch signifikant, so fand sich bei der Dauer des Ansprechens ein deutlicher Einfluss der Krankheitslast vor Infusion der Zellen: Patienten mit mehr als 5% Blasten überlebten danach median
17 Monate und waren median 6,3 Monate lang progressionsfrei, während beide Medianwerte bei denen mit minimaler Resterkrankung noch nicht erreicht sind. Die Prognose wurde weder durch die Dosierung der infundierten CAR-T-Zellen noch durch eine nachfolgende allogene Stammzelltransplantation, die bei
17 Patienten durchgeführt wurde, signifikant beeinflusst, wohl aber durch das Ausmaß der Expansion der infundierten Zellen in vivo.
Interessanterweise, so Smith, war auch die Häufigkeit von schweren Nebenwirkungen, v. a. Zytokin-Release-Syndromen (CRS) und Neurotoxizitäten von der Blastenzahl vor Gabe der manipulierten T-Zellen abhängig: Patienten mit minimaler Resterkrankung vor der Behandlung entwickelten in keinem einzigen Fall ein schweres CRS und in 19% der Fälle neurologische Nebenwirkungen; unter denen mit ≥ 5% Blasten zu Beginn traten in einem Drittel der Fälle ein schweres CRS und bei 58% Neurotoxizitäten auf. Insbesondere das neurologische Risiko korrelierte auch mit dem Ausmaß der maximalen Expansion der T-Zellen in vivo.
Diese Daten legen den Schluss nahe, so Smith, dass ein früher Einsatz der CAR-T-Zellen – vor Auftreten eines morphologischen Rezidivs – die Überlebenschancen der Patienten erhöhen könnte.
Neurologische Toxizitäten sind nach der Infusion von CAR-T-Zellen häufig, wenngleich in aller Regel reversibel. In der Phase-I-Studie am Memorial Sloan Kettering Cancer Center wurden demografische und behandlungsbedingte Faktoren, Blutparameter sowie mit der Zelltherapie zusammenhängende Faktoren auf ihren prädiktiven Wert im Hinblick auf die Neurotoxizitäten analysiert [24].
Von den 51 in der Studie behandelten Patienten entwickelten lediglich 20 gar keine neurologischen Nebenwirkungen, bei jeweils acht, zwei, 18 und drei Patienten waren diese vom Grad 1, 2, 3 oder 4. Es traten keine Grad-5-Toxizitäten und kein Fall von Hirnödem auf. Von den zahlreichen untersuchten Parametern zeigten vor allem eine hohe Krankheitslast vor der Zellinfusion (≥ 50% Blasten) und ein Zytokin-Release-Syndrom vom Grad ≥ 3 sowie einige Zytokine und das Ausmaß der CAR-T-Zell-Expansion an Tag 7 einen Zusammenhang mit der Entwicklung schwerer neurologischer Symptome. In einer multivariaten Analyse blieben letztlich nur ein Thrombozytenwert von unter 60.000/µl oder eine mittlere korpuskuläre Hämoglobinkonzentration von > 33,2% und eine morphologische Erkrankung zu Beginn (> 5% Blasten) als relevante Faktoren übrig, die mit 95% Sensitivität und 70% Spezifität das Auftreten einer schweren Neurotoxizität vorhersagen konnten.
Zumindest bei der Gabe dieses Zellpräparats, so Park, sollte es mit diesen klinischen und serologischen Biomarkern möglich sein, Risikopatienten zu identifizieren und zu versuchen, der Entwicklung oder zumindest der Exazerbation neurologischer Symptome entgegenzuwirken.
Bispezifischer Antikörper: Möglichst früh geben?
Eine weitere, bereits zugelassene immunologische Salvagetherapie-Option für Patienten mit fortgeschrittener ALL ist der bispezifische T-Cell-Engager (BiTE)-Antikörper Blinatumomab, der durch eine anti-CD3- und eine anti-CD19-Komponente CD3-positive zytotoxische T-Zellen mit den CD19-positiven leukämischen Zellen zusammenbringt. In der Phase-III-Studie TOWER konnte Blinatumomab das mediane Überleben von Patienten mit rezidivierter oder refraktärer ALL gegenüber einer konventionellen Chemotherapie von 4,0 auf
7,7 Monate beinahe verdoppeln [25]. In einer Subgruppenanalyse, die Hervé Dombret, Paris, in Madrid präsentierte, wurde untersucht, ob es eine Rolle spielt, die wievielte Salvagetherapie der bispezifische Antikörper darstellt [26].
Das Ergebnis war beeindruckend: Wurde Blinatumomab als erste Salvagetherapie gegeben, so lag die Rate an kompletten Remissionen bei 51% und die mediane Überlebensdauer bei 11,1 Monaten; hatten die Patienten vorher schon mindestens eine andere Salvagetherapie erhalten, so betrugen die entsprechenden Werte nur 39,5% bzw. 5,1 Monate. In beiden Fällen war Blinatumomab zwar der Standard-Chemotherapie deutlich überlegen, so Dombret, aber die Ergebnisse würden dafür sprechen, dass der Nutzen des bispezifischen Antikörpers umso größer ist, je früher im Krankheitsverlauf er eingesetzt wird.
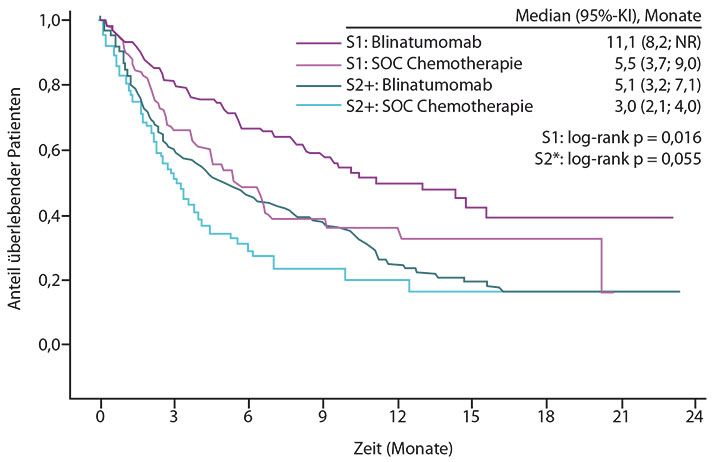
Literatur
1. Stone R et al. N Engl J Med 2017, June 23 [prepub ahead of print, DOI 10.1056/NEJMoa1614359].
2. Altman J et al. ASCO 2017, Abstract #7003.
3. Altman J et al. EHA 2017, Abstract #S110.
4. Giles F et al. Cancer 2005; 104: 547-54.
5. Levis MJ et al. ASH 2012, Abstract #643.
6. Hills R et al. EHA 2017, Abstract #S475.
7. Oran B et al. EHA 2017, Abstract #S792.
8. Stein E et al. ASCO 2017, Abstract #7004.
9. Stein E et al. EHA 2017, Abstract #S471.
10. Daver N et al. AACR 2016, Abstract #3205.
11. Yang H et al. Leukemia 2014; 28: 1280-8.
12. Daver N et al. EHA 2017, Abstract #S474.
13. ClinicalTrials.gov No. NCT02397720.
14. ClinicalTrials.gov No. NCT03092674.
15. Pratz K et al. EHA 2017, Abstract #S472.
16. ClinicalTrials.gov No. NCT02993523.
17. Wei A et al. EHA 2017, Abstract #S473.
18. ClinicalTrials.gov No. NCT03069352.
19. Venditti A et al. EHA 2017, Abstract #S111.
20. Versluis J et al. EHA 2017, Abstract #S112.
21. Gökbuget N et al. Blood 2012; 120: 2032-41.
22. Buechner J et al. EHA 2017, Abstract #S476.
23. Park J et al. EHA 2017, Abstract #S479.
24. Park J et al. EHA 2017, Abstract #S143.
25. Kantarjian H et al. N Engl J Med 2017; 376: 836-47.
26. Dombret H et al. EHA 2017, Abstract #S478.

Prof. Dr. med. Karl-Anton Kreuzer
Klinik I für Innere Medizin
Universitätsklinikum Köln
Kerpener Straße 62, 50937 Köln
+49 221 478 97626
+49 221 478 97627
Karl-anton.kreuzer[at]uni-koeln[dot]de