Sarkome – Zytostatika in neuer Verpackung, Angiogenese-Inhibitoren und Immuntherapien
ASCO 2017, Chicago
Auch bei seltenen Tumorentitäten, wie sie die Sarkome darstellen, gibt es in den letzten Jahren zunehmend Fortschritte zu vermelden. Einige zielgerichtete Therapien – vor allem solche, die sich gegen die Tumorangiogenese richten – sind bereits für verschiedene Subtypen von Weichteilsarkomen zugelassen, und beim diesjährigen Kongress der American Society of Clinical Oncology (ASCO) wurde dazu und vor allem zu Immuntherapien einiges an hoffnungsvoll stimmenden Daten präsentiert.
Klassische Zytostatika werden in immer mehr Anwendungen durch neue zielgerichtete Substanzen oder durch Biologicals wie monoklonale Antikörper ersetzt, aber völlig ausgedient haben auch sie noch nicht. Hin und wieder werden für zytotoxische Substanzen zum Beispiel neue Galeniken entwickelt, die es dem Medikament ermöglichen, sich in höheren Konzentrationen im Tumor anzureichern und so die gleiche oder sogar stärkere Wirkung mit einer geringeren systemischen Toxizität zu verbinden. Ein Beispiel dafür ist Aldoxorubicin, in dem das alte Standard-Anthrazyklin Doxorubicin über einen kovalenten chemischen Linker an Albumin gebunden wird. Die Albumin-Komponente führt zu einer selektiven Anreicherung des Konjugats im Tumorgewebe. Im sauren Milieu in der Umgebung der Tumorzellen wird die Verbindung gelöst und das Zellgift freigesetzt, das dann vorzugsweise in den Krebszellen wirkt. Bei Weichteilsarkomen, wo Doxorubicin seit jeher zu den Standardtherapien gehörte, hatte die neue Galenik in Tiermodellen und in einer Phase-IIb-Studie eine deutlich stärkere Aktivität gezeigt als die alte Substanz – ohne Anzeichen für eine kumulative kardiale Toxizität – trotz eines medianen Dosisäquivalents von 1.500 mg/m2 [1].
Deshalb wurde Aldoxorubicin nun in einer Phase-III-Studie in einer Dosierung von 350 mg/m2 in dreiwöchigen Zyklen (äquivalent zu 260 mg/m2 konventionellem Doxorubicin) randomisiert gegen eine Vergleichstherapie getestet – wobei die Prüfärzte bei der Vergleichstherapie die Auswahl aus einer ganzen Reihe von Regimes hatten: Pazopanib, Gemcitabin/Docetaxel, Dacarbazin, Doxorubicin oder Ifosfamid [2]. Eingeschlossen wurden in 79 Ländern weltweit 433 Patienten mit Weichteilsarkomen, die nach einer vorangegangenen Chemotherapie rezidiviert oder gegen sie refraktär waren. Darunter waren 42,5% Leiomyosarkome und 15% Liposarkome (d. h. 57,5% sogenannte L-Sarkome) sowie 9% Synovialsarkome und 33,5% andere Entitäten. Primärer Endpunkt war das progressionsfreie Überleben, sekundäre Endpunkte waren Ansprechrate, Krankheitskontrollrate, Gesamtüberleben und Sicherheit.
Beim progressionsfreien Überleben, so Sant Chawla, Santa Monica, fiel in der gesamten Intention-to-treat-Population die Differenz von median 4,11 Monaten unter Aldoxorubicin und 2,96 Monaten unter den Kontrolltherapien nicht signifikant aus (Hazard Ratio 0,81; p =0,0870). Interessanterweise gab es aber Unterschiede hinsichtlich Geografie und Histologie:
• Bei den Patienten aus Nordamerika und Australien war Aldoxorubicin mit median 4,21 versus 2,96 Monaten im Vorteil (Hazard Ratio 0,71; p = 0,0225), während in Europa und Lateinamerika kein signifikanter Unterschied zu finden war (median 2,96 vs. 3,02 Monate; HR 1,11; p = 0,6439).
• Bei den Patienten mit L-Sarkomen war das modifizierte Anthrazyklin ebenfalls wirksamer mit median 5,32 gegenüber 2,96 Monaten für die anderen Therapien (HR 0,62; p = 0,0070).
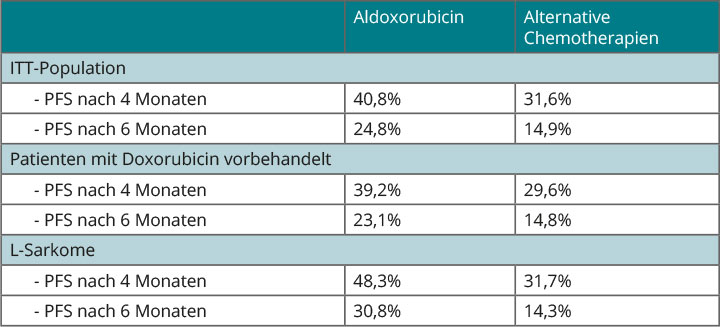
Tab. 1 zeigt, dass der – wenn auch nicht signifikante – Unterschied beim progressionsfreien Überleben nach vier und nach sechs Monaten zu sehen war, dass er unabhängig von einer bereits stattgefundenen Vorbehandlung mit Doxorubicin und am ausgeprägtesten bei den L-Sarkomen war. Beim Gesamtüberleben war im gesamten Kollektiv kein Unterschied zwischen den verschiedenen Therapiearmen zu erkennen (Aldoxorubicin median 12,88, andere Substanzen 12,16 Monate; HR 0,97; p = 0,8555). Die objektive Ansprechrate war im Verumarm mit 8,3% beinahe doppelt so hoch wie im Kontrollarm (4,2%), aber der Unterschied fiel dennoch nicht signifikant aus (p = 0,1106); bei den L-Sarkomen war der Unterschied mit 10,0% versus 4,0% noch etwas ausgeprägter und näher der Grenze zur Signifikanz (p = 0,0790). Wenn man Krankheitsstabilisierungen hinzunahm, fiel der Unterschied bei der Krankheitskontrollrate in der gesamten Population mit 33,5% versus 25,1% beinahe (p = 0,0583) und bei den L-Sarkomen deutlich signifikant aus (41,7% vs. 27,0%; p = 0,0161).
Eines der Hauptprobleme bei der Therapie mit konventionellen Anthrazyklinen ist die kardiale Toxizität, die mit der kumulativen Dosierung zunimmt. In der vorliegenden Studie, so Chawla, hatte sich gegenüber der Subgruppe von Patienten im Kontrollarm, die mit Doxorubicin behandelt wurden, der Anteil der Patienten mit einer mindestens 20%igen Reduktion der linksventrikulären Auswurffraktion unter Aldoxorubicin von 8,5% auf 3,8% mehr als halbiert. Werte von unter 50% im Verlauf der Therapie wiesen 19,1% der Patienten unter Doxorubicin, aber nur 4,2% unter Aldoxorubicin auf, obwohl die Patienten von dem neuen Anthrazyklin bis zu 40 Zyklen bekommen hatten.
Bei den nicht-kardialen Grad-3/4-Toxizitäten unterschied sich Aldoxorubicin kaum vom normalen Doxorubicin, obwohl hier insgesamt die drei- bis vierfache Dosis gegeben wurde. Auch wenn unklar bleibt, warum die europäischen und lateinamerikanischen Patienten weniger von dem neuen Anthrazyklin profitierten, sehen die Autoren wegen des längeren progressionsfreien Überlebens bei den nordamerikanischen Patienten und wegen der guten Verträglichkeit Aldoxorubicin als überlegen und als gute Alternative bei der Behandlung insbesondere von Patienten mit rezidivierten oder refraktären L-Weichteilsarkomen an.
Solitäre fibröse Tumoren: Pazopanib wirksam
Solitäre fibröse Tumoren werden in der WHO-Klassifikation unter die intermediären, selten metastasierenden fibroblastären/myofibroblastären Sarkome eingeordnet. Da sie stark vaskularisiert sind und in Tumor und umgebendem Gewebe der vaskuläre endotheliale Wachstumsfaktor (VEGF) und seine Rezeptoren exprimiert werden, wurden in einer internationalen Phase-II-Studie Patienten mit fortgeschrittenen Tumoren dieser Histologie mit dem Angiokinase-Inhibitor Pazopanib behandelt. Da bei diesen Erkrankungen auch maligne Verläufe vorkommen (für die Pazopanib zugelassen ist) und diese weniger gut auf anti-angiogene Therapien anzusprechen scheinen, wurden die Patienten in eine Subgruppe mit typischen und eine mit malignen/dedifferenzierten solitären fibrösen Tumoren stratifiziert. Beim
ASCO-Kongress stellte Javier Martin Broto, Sevilla, die Ergebnisse für die letztere Gruppe vor [3].
Im Verlauf von etwas mehr als zwei Jahren konnten in 13 Zentren in Spanien, Italien und Frankreich 34 solche Patienten eingeschlossen (31 mit maligner und drei mit dedifferenzierter Histologie) und mit 800 mg/d Pazopanib bis zur Progression oder zum Auftreten inakzeptabler Nebenwirkungen behandelt werden. Primärer Endpunkt war das Ansprechen nach den Choi-Kriterien, und von 31 auswertbaren Patienten erreichten hier 16 (52%) eine partielle Remission, sieben (22%) eine Krankheitsstabilisierung, und acht (26%) waren progredient. Nach den RECIST-Kriterien waren nur eine partielle Remission (3%), 19 Stabilisierungen (61%) und elf progrediente Erkrankungen (35%) diagnostiziert worden. Das progressionsfreie Überleben lag bei median fünfeinhalb Monaten, nach eineinhalb Jahren waren noch 72% der Patienten am Leben, aber die Überlebenschancen waren deutlich vom Ansprechen abhängig. Außerdem zeigten Patienten mit größeren Tumoren, einer höheren Mitoserate und einem dedifferenzierten Subtyp ein signifikant schlechteres progressionsfreies Überleben. In einer multivariaten Analyse waren nur die Choi-Kriterien unabhängig mit der Prognose korreliert: Patienten mit einer progressiven Erkrankung, ermittelt anhand dieser Kriterien, hatten zwölffach erhöhtes Mortalitätsrisiko (HR 11,9; p = 0,003).
In dieser ersten prospektiven Studie zu malignen solitären fibrösen Tumoren zeigte sich Pazopanib gegenüber historischen Kontrollen, die eine Chemotherapie erhalten hatten, bezüglich des Ansprechens, des progressionsfreien und des Gesamtüberlebens überlegen und etwa vergleichbar mit anderen anti-angiogenen Therapien wie Sunitinib. Die Choi-Kriterien für das Ansprechen haben sich hier als signifikanter und unabhängiger Prognosefaktor für das Überleben bewährt.
Alveoläres Weichteilsarkom: neuer Angiokinase-Inhibitor vielversprechend
Unter den Sarkomen unklarer Herkunft reiht sich das alveolare Weichteilsarkom in der WHO-Klassifikation unter den eher malignen Subtypen ein. Es ist ebenfalls sehr selten (ungefähr 0,5–1% aller Weichteilsarkome), betrifft vor allem junge Patienten und spricht schlecht auf Chemotherapie an. In Phase-II-Studien hat der Angiokinase-Inhibitor Cediranib Wirksamkeit gezeigt, die nun in einer randomisierten Phase-II-Studie überprüft wurde [4]. 48 Patienten wurden in zwölf Zentren in Großbritannien, Australien und Spanien im Verhältnis 2 : 1 randomisiert, doppelblind Cediranib (30 mg/d) oder Placebo zu erhalten. Primärer Endpunkt war das prozentuale Ansprechen bestimmter Marker-Läsionen bis Woche 24, und hier zeigte sich Cediranib mit einer Abnahme um 8,3% dem Placebo überlegen, unter dem die Tumoren um 13,4% an Größe zunahmen, so Ian Judson, London (p = 0,0010). Auch beim progressionsfreien Überleben schnitt der Kinase-Inhibitor mit median 10,8 versus 3,7 Monaten und einer 12-Monats-Rate von 47,7% versus 22,5% zumindest numerisch besser ab (HR 0,58; p = 0,059), ebenso beim Gesamtüberleben (nach zwölf Monaten 94% vs. 66%; HR 0,66; p = 0,41).
An Nebenwirkungen vom Grad 3 oder höher wurden unter Cediranib vor allem ein Hypertonus, eine GGT-Erhöhung, Diarrhö und Asthenie/Fatigue gesehen; sie waren mit Dosisreduktionen und Antihypertensiva gut handhabbar.
Regorafenib bei Ewing-Sarkomen
Auch die histologische Herkunft der Ewing-Sarkome ist nicht geklärt. In einer einarmigen, multizentrischen Phase-II-Studie testeten US-amerikanische Kollegen den Kinase-Inhibitor Regorafenib bei insgesamt 30 Patienten mit Ewing-Sarkomen und verwandten Tumoren von Weichgewebe und Knochen, die vorher mindestens eine Therapie erhalten hatten; darunter durften keine oralen Kinase-Inhibitoren sein [5]. Primärer Endpunkt war das progressionsfreie Überleben nach acht Wochen, wobei ein Unterschied von 50% gegenüber 25% mit statistischer Signifikanz nachgewiesen werden sollte, so Steven Attia, Jacksonville. Am Ende lag der Wert bei 73%, womit die Wirksamkeit von Regorafenib in dieser Indikation zweifelsfrei belegt ist. Bezüglich des Ansprechens wurden drei partielle Remissionen neben 18 Krankheitsstabilisierungen gesehen, die mediane Dauer des Ansprechens lag bei 5,5 Monaten, das mediane Gesamtüberleben ist noch nicht erreicht. Bei der Toxizität von Regorafenib gab es keine neuen Signale.
Angesichts der Tatsache, dass die Patienten im Median fünf Vortherapien erhalten hatten, ist diese Wirkung bemerkenswert, so Attia. Die Rekrutierung für die Studie geht weiter, ebenso die Untersuchung des Tumorgewebes, von dem man sich Aufschlüsse darüber erhofft, welche Patienten besonders von dem Inhibitor profitieren könnten.
Impfung für dendritische Zellen gegen Sarkome
Das Tumorantigen NY-ESO-1 wird auf einer Reihe von Weichteilsarkomen exprimiert, was US-amerikanische Kollegen zu einer neuen Immuntherapie inspirierte: In dem CMB305 genannten Präparat kombinieren sie einen Lentivirus-Vektor, der dendritische Zellen infiziert, NY-ESO-1 exprimiert und dadurch die Induktion und Expansion von für das Antigen spezifischen T-Zellen bewirkt, mit einem TLR-4-Agonisten, der die Immunogenität der infizierten dendritischen Zellen verstärken und die Ausschüttung von Anti-NY-ESO-1-Antikörper bewirken soll. In der Phase-I-Studie, die Neeta Somaiah, Houston, in Chicago vorstellte, erhielten bisher 25 vorbehandelte Patienten (darunter 15 Synovialsarkome und acht myxoid/rundzellige Liposarkome) bis zu ein Jahr lang Injektionen des Vektors und des Agonisten [6].
Das Präparat scheint gut verträglich zu sein; 64% der Patienten entwickelten spezifische T-Zellen und 72% Antikörper gegen NY-ESO-1. Bei den Patienten mit Synovialsarkom wurden in 53% der Fälle eine Krankheitsstabilisierung beobachtet, bei den übrigen in 75% (nach immunologischen Ansprechkriterien). Nach drei Monaten waren noch drei Viertel der Patienten progressionsfrei am Leben, die Überlebensraten nach einem Jahr betrugen 86% für die Patienten mit Synovialsarkomen und 100% für die anderen.
Die Autoren halten angesichts dieser Resultate eine randomisierte Studie für angezeigt, um die Substanzen bei Weichteilsarkomen weiter zu untersuchen. In einer randomisierten Phase-II-Studie wird CMB305 außerdem zurzeit in den USA bereits in Kombination mit dem PD-L1-Checkpoint-Inhibitor Atezolizumab gegen Atezolizumab alleine getestet [7].
Checkpoint-Inhibitoren haben auch bei Sarkomen Potenzial
Auch die inzwischen schon beinahe "klassischen" Immuntherapien mit Checkpoint-Inhibitoren werden bei Sarkomen untersucht: In einer Phase-II-Studie wurden etwa 85 Patienten mit metastasierten Sarkomen verschiedenster Histologien, die auf keine vorangegangenen Therapien mehr angesprochen hatten, mit dem PD-1-Antikörper Nivolumab entweder alleine oder in Kombination mit dem CTLA-4-Antikörper Ipilimumab behandelt [8]. 58% der Patienten hatten sich gegen mindestens drei andere Therapien als refraktär erwiesen.
Von 38 auswertbaren Patienten im Nivolumab-Monotherapiearm, so Sandra D´Angelo, New York, sprachen drei (5%) mit einer partiellen Remission und 15 (39%) mit einer Krankheitsstabilisierung an. Bei den 38 Patienten, die die Kombination der beiden Inhibitoren erhalten hatten, wurden zwei komplette (5%) und fünf partielle Remissionen (13%) sowie 19 Stabilisierungen (50%) registriert. Das mediane progressionsfreie Überleben in den beiden Gruppen wird mit 2,1 bzw. 4,4 Monaten angegeben, das mediane Gesamtüberleben mit 10,7 bzw. 14,3 Monaten. Eine Überlebensrate von 54% nach einem Jahr in der Kombinationskohorte übertraf die Erwartungen in diesem stark vorbehandelten Kollektiv, so D´Angelo, sodass weitere Studien dringend wünschenswert wären.
In einer multizentrischen US-amerikanischen Phase-II-Studie schließlich wurde der andere PD-1-Antikörper, Pembrolizumab, bei insgesamt 40 Patienten mit Weichteilsarkomen verschiedener Histologien sowie bei 40 Patienten mit Knochensarkomen (22 Osteo-, 13 Ewing- und fünf Chondrosarkome) getestet [9]. Die Behandlung erwies sich als wirksam vor allem bei undifferenzierten pleomorphen (eine Komplett- und drei partielle Remissionen) und bei dedifferenzierten Liposarkomen (zwei partielle Remissionen), sodass von diesen beiden Histologien noch einmal je 30 Patienten in eine Expansionskohorte eingeschlossen werden sollen, so Melissa Burgess, Pittsburgh. Im Übrigen schien das Ansprechen mit der PD-L1-Expression im Tumorgewebe zu korrelieren.
Literatur
1. Chawla SP et al. JAMA Oncol 2015; 17: 1-9.
2. Chawla SP et al. ASCO 2017, Abstract #11000.
3. Martin Broto J et al. ASCO 2017, Abstract #11003.
4. Judson IR et al. ASCO 2017, Abstract #11004.
5. Attia S et al. ASCO 2017, Abstract #11005.
6. Somaiah N et al. ASCO 2017, Abstract #11006.
7. ClinicalTrials.gov No. NCT02609984.
8. D´Angelo SP et al. ASCO 2017, Abstract #11007.
9. Burgess MA et al. ASCO 2017, Abstract #11008.

Prof. Dr. Clemens-Martin Wendtner
Klinik für Hämatologie, Onkologie, Immunologie, Palliativmedizin, Infektiologie und Tropenmedizin,
Klinikum Schwabing
Akademisches Lehrkrankenhaus der
Universität München
Kölner Platz 1, 80804 München
+49 89 3068 2228
+49 89 3068 3912
E-Mail schreiben