Artikel als PDF downloaden.
Myeloproliferative Neoplasien – neue Daten zur Genetik und neue Therapien
ASH 2015
Vor kaum mehr als zehn Jahren begann mit der Entdeckung der Punktmutation V617F in der Janus-Kinase 2 (JAK2; [1]) eine Erfolgsgeschichte, die noch lange nicht zu Ende ist. Diese Driver-Mutation findet sich bei über der Hälfte der Patienten mit primärer Myelofibrose und, wie man mittlerweile weiß, bei einem noch höheren Anteil derjenigen mit essenzieller Thrombozythämie (ET) sowie bei fast allen Patienten mit Polycythaemia vera (PV). Mit der Entdeckung der JAK2-Mutation wurde es möglich, spezifische Inhibitoren zu entwickeln, von denen der erste, Ruxolitinib, nach erstaunlich kurzer Zeit Eingang in die Klinik fand. Wie bei der Jahrestagung 2015 der American Society of Hematology (ASH) in Orlando deutlich wurde, befindet sich das Feld der BCR/ABL-negativen myeloproliferativen Neoplasien weiter in rascher Expansion, was sowohl die Erforschung des genetischen Hintergrunds als auch die Entwicklung neuer Therapieoptionen betrifft.
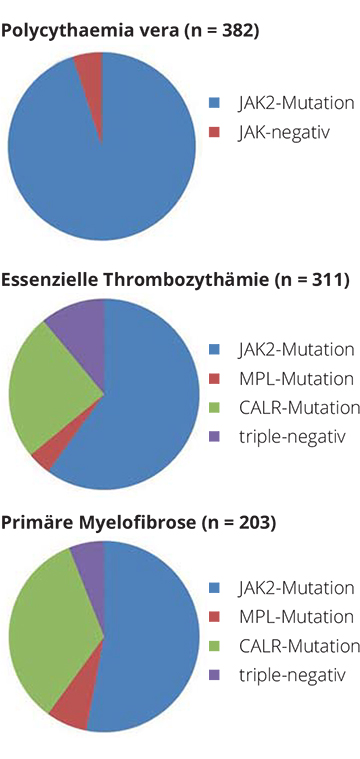
Neben der JAK2-Mutation wurden bisher zwei weitere Treibermutationen im Calreticulin-Gen (CALR) und im MPL-Gen gefunden. Bei der überwiegenden Mehrzahl der Patienten mit primärer Myelofibrose, ET und PV findet sich eine dieser drei Veränderungen ([2]; Abb. 1). Eine Reihe weiterer somatischer Veränderungen (etwa in TET2, CBL, EZH2, DNMT3A, ASXL1 usw.) können hingegen zusätzlich vorliegen und sind nicht spezifisch für die BCR/ABL-negativen myeloproliferativen Erkrankungen, sondern kommen auch bei anderen hämatologischen Störungen wie etwa den Myelodysplasien vor.
Um mehr Licht in die Rolle dieser anderen Mutationen zu bringen, untersuchte die Arbeitsgruppe an der Mayo Clinic in Rochester unter Alayew Tefferi mithilfe von Next Generation Sequencing (NGS) ein Panel von 27 Genen bei Patienten mit primärer Myelofibrose [3]. Von den 180 Patienten mit primärer Myelofibrose in dieser Studie waren 9% triple-negativ, d. h. sie hatten keine Mutation in JAK2, CALR oder MPL. Der Karyotyp war bei 40% abnormal und bei 12% ungünstig. 83% dieser Patienten wiesen Non-Driver-Mutationen in unterschiedlicher Anzahl auf, und je mehr solche Mutationen gleichzeitig vorhanden waren, desto schlechter war die Prognose. Einzelne Mutationen mit der schlechtesten Prognose waren solche in ASXL1, SRSF2, CBL und RUNX1. In einer weiteren aufwendigen NGS-Analyse konnten die Mayo-Forscher zeigen [4], dass Non-Driver-Mutationen auch bei 40% der Patienten PV und ET auftreten und auch hier prognostische Bedeutung bezüglich der Überlebenszeit haben.
Bedeutung der Knochenmarkfibrose nicht unterschätzen
Eine italienische Studie stellte die provokante Frage, ob die Knochenmarkfibrose wirklich ein negativer Prädiktor ist [5] – das wurde bisher noch nie wirklich detailliert untersucht. Die deutschen Arbeiten von Thiele und Kvasnicka zeigten [6], dass die frühe Fibrose vom Grad 1 mit erhöhter Letalität assoziiert ist, für die höheren Grade gibt es aber kaum Daten. Die italienischen Kollegen analysierten jetzt das Knochenmark von 540 Patienten mit primärer Myelofibrose zum Zeitpunkt der Diagnose und führten zusätzlich ein Mutations-Screening auf Driver- und ausgewählte Non-Driver-Mutationen durch (EZH2, ASXL1, IDH1/2, SRSF2); der Fibrose-Grad wurde nach dem European Consensus Scoring System (MF-0 bis MF-3) festgelegt.
Die Patienten wurden median 3,7 Jahre lang beobachtet, bisher sind 34,1% von ihnen verstorben. MF-0 tritt bei 9,3% der Patienten auf, MF-1 bei 33,3%, MF-2 bei 36,3%, MF-3 bei 21,1%, und je höher der Fibrose-Grad, desto mehr konstitutionelle Symptome lagen vor, desto größer war die Milz und desto ausgeprägter Anämie, Thrombozytopenie und IPSS-Risiko-Score. Für den erfahrenen Kliniker mag das trivial klingen, aber es wurde zum einen noch nie in einem so großen Kollektiv belegt und könnte andererseits bedeuten, dass die Knochenmark-Diagnostik auch wieder in die Prognose-Systeme Einzug hält. Triple-negative Patienten haben interessanterweise nicht notwendigerweise eine stärkere Fibrose, aber die Zahl molekularer Hochrisikofaktoren – Mutationen in ASXL1, EZH2 usw. – korreliert sehr wohl mit dem Fibrose-Grad (Abb. 2).
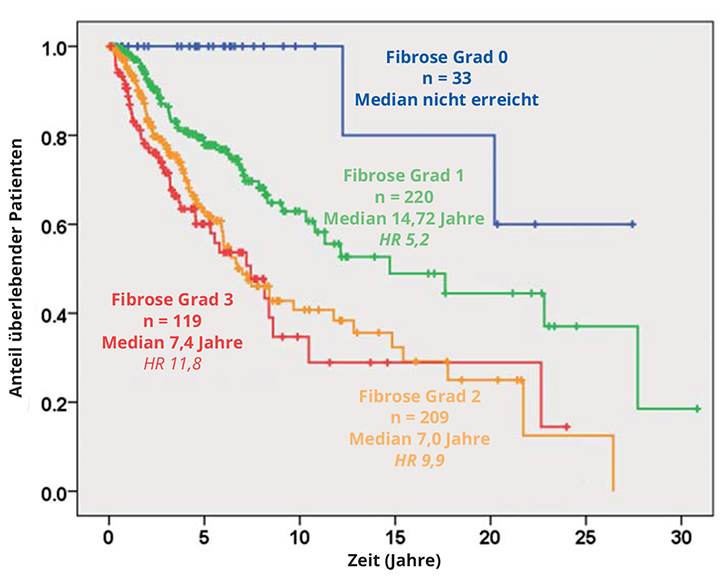
Fazit dieser klinisch sehr wichtigen Studie ist, dass die Knochenmark-Biopsie nicht nur diagnostische, sondern auch prognostische Bedeutung hat: Die Fibrose korreliert ganz klar mit definierten klinischen und molekularen Variablen, diese haben einen unabhängigen Einfluss auf das Überleben, und man muss sich überlegen, ob man die Knochenmarkfibrose in prognostische Scores einbringt.
Einfluss von Non-Driver-Mutationen
Die allogene Stammzelltransplantation ist bei der Myelofibrose trotz aller modernen medikamentösen Entwicklungen immer noch das einzige kurative Verfahren. Auch hier war unklar, welchen Einfluss die molekularen Veränderungen auf das Ergebnis haben. Nicolaus Kröger, Hamburg, analysierte 169 Patienten mit v. a. primärer Myelofibrose, die im Alter von bis zu 75 Jahren v. a. von nicht verwandten Spendern und mit einer Konditionierung reduzierter Intensität transplantiert worden waren [7].
Nur elf der 169 Patienten hatten keine Mutation, eine Mutation war bei 41%, zwei bei 30%, drei bei 11,4% und vier bei 5% nachweisbar. Die günstigsten Faktoren für das Ergebnis nach Transplantation sind – nicht sehr überraschend – jüngeres Alter (< 58 Jahre; p < 0,01), ein niedriger DIPSS-Risiko-Score (p = 0,002), das HLA-Matching (p = 0,04) sowie –das ist neu – das Vorliegen einer CALR-Mutation (p = 0,005; Tab. 1).
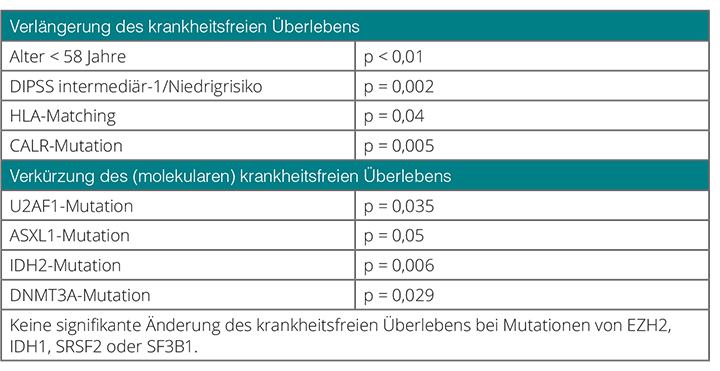
Primäre Motivation für diese Studie war, den Einfluss der Non-Driver-Mutationen aufzuklären. Hier gibt es Faktoren, die einen Einfluss auf das krankheitsfreie Überleben haben, darunter U2AF1, ASXL1, IDH2 und DNMT3A (Tab. 1); keine signifikanten Änderungen zeigen sich hingegen bei Mutationen von EZH2, IDH1, SRSF2 und SF3B1. Erstaunlicherweise stellt SRSF2, das eigentlich mit einer schlechten Prognose assoziiert wird, nach Transplantation offenbar kein Problem mehr dar. Möglicherweise kann also die Transplantation den negativen Prädiktor SRSF2 auf irgendeine Art und Weise neutralisieren.
Ruxolitinib bei Myelofibrose auch nach fünf Jahren im Vorteil
Ruxolitinib ist der erste und bisher einzige JAK-Inhibitor, der zur Therapie der Myelofibrose und der Polycythaemia vera zugelassen ist. Zur Myelofibrose-Therapie gab es in Orlando die 5-Jahres-Daten der COMFORT-II-Studie, die von Claire Harrison, London, vorgestellt wurden [8]. Ruxolitinib war hier mit der besten sonst zum Zeitpunkt des Studienbeginns verfügbaren Therapie (BAT) nach Ansicht des jeweiligen Prüfarztes verglichen worden, in der COMFORT-I-Studie hingegen mit Placebo. In beiden Studien war es zu einem hochsignifikanten Rückgang der Splenomegalie, einer Besserung der klinischen Symptome, vor allem aber auch zu einer Verlängerung der Überlebenszeit gekommen.
So hatte COMFORT II für Ruxolitinib einen Rückgang der Milzgröße um ≥ 35% bei 28% der Patienten gegenüber keinem einzigen unter BAT gezeigt. Dieser Unterschied hatte nach drei Jahren bei guter Verträglichkeit von Ruxolitinib angehalten; die Patienten durften von der BAT- in die Ruxolitinib-Gruppe wechseln, wenn während der Initialphase das Milzvolumen um 25% zugenommen hatte. Nach median 4,3 Jahren Nachbeobachtungszeit zeigte sich nun, dass nach fünf Jahren noch 50 Patienten mit Ruxolitinib behandelt werden, davon 39 der 146 in der ursprünglichen Ruxolitinib-Gruppe und elf nach Cross-over aus dem BAT-Arm. Gründe für einen Abbruch im Ruxolitinib-Arm waren Nebenwirkungen (24%) und Progression (21,9%); die Nebenwirkungen traten median nach 2,6 Jahren auf. Für den Kliniker besonders interessant sind eine geringere Leukämie-Rate im Ruxolitinib-Arm (5,5% versus 6,8%) und eine geringere Mortalität (40,4% vs. 47,9%). Die mediane Überlebensdauer ist im Ruxolitinib-Arm noch nicht erreicht, im BAT-Arm liegt sie bei 4,1 Jahren (HR 0,67; p = 0,06). Der positive Überlebenseffekt von Ruxolitinib wird natürlich ein wenig verwischt durch das Cross-over von Patienten von BAT zu Ruxolitinib.
Interferon: Immer noch eine Option
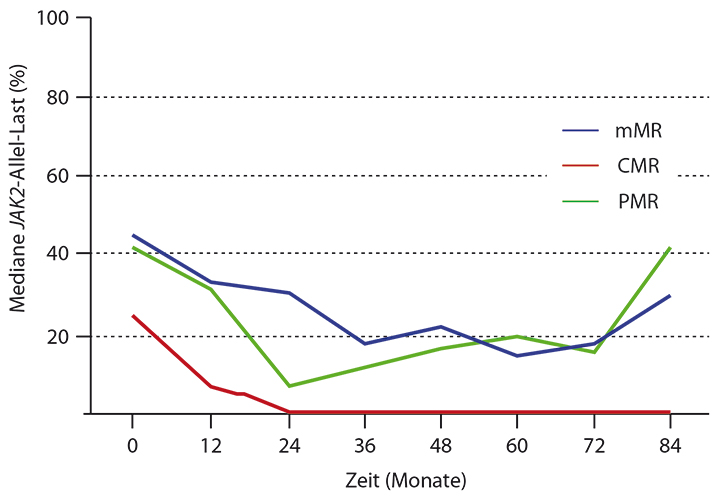
Bei der Myelofibrose wird immer wieder die initiale Gabe von Interferon diskutiert. Eine Studie aus dem M. D. Anderson Cancer Center zeigte bei PV und ET deutliche hämatologische und molekulare Remissionen, d. h. auch einen Rückgang der Allel-Last, und zu dieser Phase-II-Studie konnten in Orlando immerhin 7-Jahres-Langzeitdaten präsentiert werden [9]. Die 83 Patienten mit Behandlungsbedürftiger PV oder ET, die zu gut einem Drittel vorbehandelt waren, erhielten bemerkenswert hohe Dosierungen von pegyliertem Interferon, nämlich im Median 180 µg/Woche. Das vertrugen erwartungsgemäß nicht alle Patienten – bei mehr als einem Drittel der Patienten lag die Eingangsdosis bei 90 µg/Woche, und es gab zahlreiche Dosismodifikationen.
Vom ersten guten Ansprechen bis zum jetzigen Analysezeitpunkt nach median 87 Monaten ging die hämatologische Responserate von 79% auf 39% zurück, die molekulare von 63% auf 45%. Das ist nach sieben Jahren immer noch bemerkenswert, aber es kommt offensichtlich zu einem Wirkungsverlust. Wenn Patienten eine komplette molekulare Remission erzielen, hält diese lange an. Das entspricht auch der klinischen Erfahrung, dass es Patienten gibt, die exzellent auf Interferon ansprechen und dann auch erheblich davon profitieren. Bei denjenigen mit weniger guter molekularer Remission kommt es hingegen irgendwann wieder zu einer Erhöhung der Allel-Last.
Das molekulare Ansprechen auf pegyliertes Interferon ist also für über ein Drittel der Patienten exzellent, allerdings stellt die Toxizität weiterhin ein Problem dar, das in der Langzeittherapie berücksichtigt werden muss.
Hämatologen aus Dänemark, wo man sich schon lange mit Interferon beschäftigt, testeten eine Kombination aus Interferon und Ruxolitinib bei 30 Patienten mit myeloproliferativen Neoplasien (sieben mit präfibrotischer oder hyperproliferativer primärer Myelofibrose, 20 mit Polyzythämie, drei mit Post-PV-Myelofibrose; [10]): Sie erhielten pegyliertes Interferon mit 45 µg/Woche (Pegasys) bzw. 35 µg/Woche (PegIntron) plus zweimal 20 mg/d Ruxolitinib. Response-Kriterien waren die palpierte Milzgröße, die JAKL2V617F-Allel-Last und das Nebenwirkungsprofil. Bei einer Therapiedauer von bisher 24 Wochen brachen nur drei Patienten die Behandlung wegen Nebenwirkungen ab. Bemerkenswerterweise waren 27 Patienten bereits mit Interferon vorbehandelt, das von 18 nicht vertragen worden war und bei fünf nicht gewirkt hatte. Dennoch konnte mit dieser Kombination bei den PV-Patienten, die vorher phlebotomiert werden mussten, in 78% der Fälle schon ab der vierten Woche der Hämatokrit ohne Phlebotomie kontrolliert werden. Das hämatologische Ansprechen war sehr gut mit 63,3% kompletten und 26,7% partiellen Remissionen; nur 10% der Patienten blieben ohne Response. Auch die JAK2-Allel-Last ging schon nach wenigen Wochen deutlich zurück.
Die Kombination ist also gut wirksam und verträglich (bei einigen SAEs). Kontrovers wurde diskutiert, ob Ruxolitinib in Monotherapie nicht einen ähnlichen Erfolg gebracht hätte. Bemerkenswert ist auf jeden Fall, dass die JAK-Allel-Last sehr schnell abgenommen hat – nach nur wenigen Wochen.
Kombinationen von Ruxolitinib mit Azacitidin, …
Derzeit werden zahlreiche verschiedene Kombinationen aus Ruxolitinib und für andere Indikationen zugelassenen oder neuen, noch in der Entwicklung befindlichen Substanzen bei myeloproliferativen Neoplasien erprobt. Kollegen am M. D. Anderson Cancer Center beschäftigten sich mit einer Untergruppe dieser Erkrankungen, nämlich mit Patienten mit MPN/MDS-Erkrankungen [11], die sie mit dem epigenetisch wirksamen und für bestimmte Formen von MDS zugelassenen Azacitidin in Kombination mit Ruxolitinib behandelten. Nach drei vierwöchigen Zyklen Ruxolitinib (15 mg/d) erfolgte eine sequenzielle Gabe von Azacitidin (25 mg/m2 an Tag 1–5, mit eventueller Steigerung auf bis zu 75 mg/m2). Viele Patienten waren vorbehandelt, die Hälfte litt an einer Splenomegalie, fast drei Viertel an einer Knochenmarkfibrose.
Etwa die Hälfte aller Patienten sprach nach den MDS/MPN-IWG-Kriterien an [12], bei denen mit palpabler Milz waren es 73%, hinsichtlich der Symptome (Total Symptom Score) profitierte ein Viertel der Patienten, bei Anämien 15% und im Knochenmark 45%. Das Viertel der Patienten, das JAK-positiv war mit einer medianen Allel-Last von über 40%, reagierte besonders gut auf diese Kombination. Das Gesamtüberleben lag im Median bei 15,1 Monaten, 13 Patienten mussten die Therapie abbrechen, sieben verstarben. Diese Kombination scheint gut verträglich und wirksam zu sein mit einer Response-Rate von 50%, und v. a. JAK-positive Patienten sprechen besonders gut an. Diese Kombination sollte weiter in Studien evaluiert werden.
… Sonidegib, …
Zu einer Studie, in der Ruxolitinib mit Sonidegib kombiniert wurde, gab es ein Update [13]. Der Smoothened-Inhibitor blockiert den Hedgehog-Signalweg und bewirkte im Mausmodell in Kombination mit Ruxolitinib sowohl eine Verbesserung der Splenomegalie als auch einen Rückgang der Knochenmarkfibrose im Vergleich zur Ruxolitinib-Monotherapie. 27 Patienten mit primärer (n = 16) oder sekundärer Myelofibrose (4 Post-PV, 7 Post-ET), vornehmlich mit Hoch- und Intermediär-Risiko und palpabler Splenomegalie ohne Vorbehandlung mit einem JAK-Inhibitor, erhielten Sonidegib mit 400 mg einmal täglich und Ruxolitinib mit 20 mg zweimal täglich. Endpunkt war die Reduktion der Milzgröße in MRT und CT nach 24 und 48 Wochen.
20 Patienten, also immerhin etwa drei Viertel, sind noch in der Studie mit einer Therapiedauer von 28 Wochen. Eine mehr als 35%ige Reduktion der Milzgröße in der dreidimensionalen Darstellung wurde bei zwölf von ihnen erreicht (44,4%) – das ist sehr gut, verglichen mit etwa 30% mit einer Ruxolitinib-Monotherapie. Einen nicht-palpablen Milzstatus erreichten 55% der Patienten. Der Rückgang der Allel-Last um 9% war nicht so eindrucksvoll, aber die besten Werte lagen bei immerhin 56% Rückgang. Einige Patienten erreichten eine Verbesserung (n = 2) oder Stabilisierung (n = 8) der Knochenmarkfibrose.
Die Kombination ist also wirksam und führt vor allem zu einer deutlichen Abnahme der Splenomegalie. Sie scheint auch gut verträglich zu sein, so Vikas Gupta, Toronto, weil immerhin 74% der Patienten nach dieser langen Zeit noch in der Studie sind.
… Pomalidomid …
In einer deutschen Phase-Ib/II-Studie, die Frank Stegelmann, Ulm, vorstellte [14], wurde eine Kombination aus Ruxolitinib und Pomalidomid bei Patienten mit Myelofibrose untersucht. Bei der Myelofibrose sieht man das Phänomen einer klinischen Trias aus konstitutionellen Symptomen, Splenomegalie und Zytopenien, insbesondere Anämie und Thrombozytopenie. Die beiden ersten Parameter sprechen auf Ruxolitinib sehr gut an, aber die Zytopenien bleiben ein Problem. Um hier ein neues Element in die Therapie einzuführen, wurde Pomalidomid als Kombinationspartner von Ruxolitinib gewählt, weil es Daten gibt, wonach die immunmodulatorische Therapie mit IMiDs Anämie und Thrombozytopenie in Untergruppen von Myelofibrose-Patienten bessert [15].
Einschlusskriterien für die bisher behandelten 30 Patienten waren daher das Vorliegen einer Anämie (Hb < 10 g/dl) bzw. einer Transfusionsabhängigkeit bei Patienten mit primärer, aber auch mit sekundärer Myelofibrose. Ruxolitinib wurde mit 10 mg zweimal täglich dosiert, Pomalidomid mit 0,5 mg/d und je nach Verlauf individuell angepasst. Primäre Endpunkte waren die Ansprechrate nach den IWG-MRT-Kriterien nach zwölf Zyklen und ein Rückgang der Transfusionsabhängigkeit.
Zehn der Patienten waren bereits vorbehandelt, der mediane Hb-Wert bei Studieneintritt lag bei 8,6 g/dl, sechs Patienten waren transfusionsabhängig, die mediane Milzgröße betrug 18 cm. Jeder dritte Patient hatte einen DIPSS-Score von intermediär-1, 40% von intermediär-2 und 28% einen Hochrisiko-Score, d. h. es handelte sich insgesamt eher um ein Hochrisiko-Kollektiv.
An Nebenwirkungen wurden vor allem Anämien, muskuloskelettale Schmerzen und konstitutionelle Symptome gesehen, aber die Ruxolitinib-Dosis musste selten modifiziert werden, bei immerhin fast der Hälfte der Patienten konnte sie von 10 auf 15–20 mg gesteigert werden. Es wurden bei 24 Patienten im Median acht Behandlungszyklen à 28 Tage durchgeführt, und über die Hälfte ist noch in der Studie. Bei sechs Patienten war eine klare klinische Besserung entweder durch Rückgang der Splenomegalie, durch einen Anstieg des Hb-Wertes oder einen Anstieg von sowohl Hb-Wert als auch Neutrophilen-Zahlen zu sehen. In keinem Fall kam es unter dieser Kombination zu einer Progression der Erkrankung. In 13 Fällen traten schwere Nebenwirkungen auf, die aber meist nicht Therapie-assoziiert waren, abgesehen von einem Fall einer schweren Grad-4-Anämie, einer Hepatotoxizität und einer Neuropathie vom Grad 3, die jedoch reversibel war. Abgebrochen wurde die Behandlung in 12 Fällen.
… und PRM-151
Eine interessante Neuentwicklung auf dem Gebiet der Myelofibrose ist das Medikament PRM-151, eine rekombinante Form des endogenen Plasma-Proteins Pentraxin-2, das die Differenzierung von Makrophagen induzieren kann und der Fibrose vorbeugen oder diese sogar rückgängig machen soll. Ziel einer Phase-I/II-Studie, die Srdan Verstovsek, Houston, vorstellte, war die Evaluierung von Verträglichkeit und Wirksamkeit von PRM-151 in Monotherapie oder in der Kombination mit Ruxolitinib [16]. 27 Patienten mit primärer oder sekundärer Myelofibrose vom
Grad 2 oder 3 wurden eingeschlossen, da unter anderem der Rückgang der Fibrose (um mindestens einen Grad nach den WHO-Kriterien) untersucht werden sollte. PRM-151 wurde wöchentlich oder alle vier Wochen gegeben, alleine oder in Kombination mit Ruxolitinib über insgesamt 24 Wochen. Dann wurde der klinische Nutzen evaluiert (Rückgang der Fibrose, Verbesserung von Anämie, Thrombopenie, Symptome, Milzgröße).
Nebenwirkungen waren in der von Verstovsek in Orlando vorgestellten 72-Wochen-Auswertung von 13 Patienten vor allem Fatigue, Nausea, Fieber, Husten usw., überwiegend vom Grad 1, selten höher, d. h. die Substanz war auch in der Langzeit-Phase sehr gut verträglich. Ein Rückgang der Fibrose nach den WHO-Kriterien um mindestens einen Grad war bei 54% der Patienten zu beobachten, wenn eine Computer-assistierte Methode angewendet wurde, sogar bei 85%.
Der Hb-Wert stieg von Woche 48 bis 72 an von median 8 auf über 10 g/dl, und im Gegenzug nahmen die Transfusionen deutlich ab. Ein ganz ähnliches Bild zeigte sich für die Thrombozyten, nämlich ein Anstieg von etwas über 35.000/µl auf Werte über 50.000/µl und auch hier ein ausgeprägter Rückgang der Transfusionen. Bis auf einen Patienten wiesen alle eine mehr oder weniger deutliche Symptombesserung auf, und die Splenomegalie ging bei den Patienten, bei denen eine solche vorlegen hatte, um bis zu 100% zurück.
In der Langzeitbehandlung erweist sich PRM-151 also als sehr gut verträglich und effektiv bei Patienten, die in der Anfangsphase ansprechen. Aufgrund der geringen Patientenzahlen lässt sich noch nicht sagen, ob die Kombination mit Ruxolitinib einen zusätzlichen Nutzen bringt.
Pacritinib, ein neuer JAK-Inhibitor
Alessandro Vannucchi, Florenz, beschäftigte sich mit dem neuen JAK-Inhibitor Pacritinib, der in der Phase-III-Studie PERSIST-1 bei 327 Patienten mit Myelofibrose und Intermediär-1- bis Hochrisiko-DIPSS, palpabler Milz, einem Symptom-Score von ≥ 13 und ohne vorabgegangene Therapie mit einem JAK-Inhibitor in einer 2 : 1-Randomisierung mit der besten sonst verfügbaren Therapie (BAT) verglichen wurde – in einer Dosierung von 400 mg/d [17]. Im Gegensatz zu den COMFORT-Studien hatte hier ein Drittel der Patienten Thrombozytenzahlen von < 100.000/µl und 16% sogar von < 50.000/µl, wie das bei fortgeschrittener Myelofibrose häufiger zu sehen ist; drei Viertel der Patienten waren JAK-positiv. Pacritinib ist ein Inhibitor, der neben JAK2 zum Beispiel auch die Flt3-Rezeptorkinase sowie einige weitere Faktoren hemmt. Der Vorteil scheint darin zu bestehen, dass es hier zu einer weniger ausgeprägten Myelosuppression kommt, sodass insbesondere Patienten mit Thrombozytopenie oder Anämie möglicherweise besser behandelt werden können. In Orlando präsentierte Vannucchi Subgruppenanalysen zu einzelnen Parametern, die das Outcome positiv oder negativ beeinflussen.
Im Gesamtkollektiv von PERSIST-1 zeigte sich ähnlich wie in den COMFORT-Studien eine signifikante Verkleinerung der Milz unter Pacritinib gegenüber BAT bei rund 20% der Patienten gegenüber 5% unter BAT. In einer multivariaten Analyse wurden Faktoren identifiziert, die diese Milzgröße und Symptomlast positiv oder negativ beeinflussen. Es fanden sich drei Faktoren, die die Entwicklung der Milzgröße unter Pacritinib positiv zu beeinflussen schienen:
- Bei einer Thrombozytenzahl von < 50.000/µl kann man offenbar nicht nur mit Pacritinib behandeln, sondern es gibt hier die höchsten Ansprechraten gegenüber dem Kontrollarm in Höhe von 22,9%.
- Auch bei den JAK-negativen Patienten war der Anteil zusätzlich ansprechender Patienten mit 23% ähnlich hoch, und
- auch junge Patienten (< 65 Jahre) scheinen besser zu profitieren (+ 21%).
Pacritinib führt zu einer Reduktion von Milzgröße (und auch Symptom-Score) unabhängig von unterschiedlichen Baseline-Kriterien wie Thrombopenie, Alter etc. Pacritinib ist in allen Subgruppen und bezüglich beider Endpunkte der BAT überlegen ähnlich wie Ruxolitinib in den COMFORT-Studien.
Literatur
1. Kralovics R et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779-90.
2. Klampfl T et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013; 369: 2379-90.
3. Tefferi A et al. ASH 2015, Abstract #350.
4. Tefferi A et al. ASH 2015, Abstract #354.
5. Guglielmelli P et al. ASH 2015, Abstract #351.
6. Thiele J, Kvasnicka M. Grade of bone marrow fibrosis is associated with relevant hematological findings-a clinicopathological study on 865 patients with chronic idiopathic myelofibrosis. Ann Hematol 2006; 85: 226-32.
7. Kroeger N et al. ASH 2015, Abstract #352.
8. Harrison CN et al. ASH 2015, Abstract #59.
9. Masarova L et al. ASH 2015, Abstract #60.
10. Mikkelsen SU et al. ASH 2015, Abstract #824.
11. Daver N et al. ASH 2015, Abstract #823.
12. Savona et al. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood 2015; 125: 1857-65.
13. Gupta V et al. ASH 2015, Abstract #825.
14. Stegelmann F et al. ASH 2015, Abstract #826.
15. Tefferi A et al. Pomalidomide is active in the treatment of anemia associated with myelofibrosis. J Clin Oncol 2009; 27: 4563-9.
16. Verstovsek S et al. ASH 2015, Abstract #56.
17. Vannucchi AM et al. ASH 2015, Abstract #58.

Prof. Dr. Martin Griesshammer
Klinik für Hämatologie, Onkologie und
Palliativmedizin, Mühlenkreiskliniken
Johannes Wesling Klinikum Minden
Hans-Nolte-Straße 1, 32429 Minden