Artikel als PDF downloaden.
Chronische myeloische Leukämie
ASH 2015
Die chronische myeloische Leukämie ist eine Art Schrittmacher-Krankheit der Hämatologie, wenn nicht der gesamten Onkologie: Zu ihrer Behandlung wurde mit dem Tyrosinkinaseinhibitor (TKI) Imatinib die erste zielgerichtete Substanz eingeführt, und bei ihr wurde auch zuerst die Messung der minimalen Resterkrankung zur Prognoseabschätzung in der klinischen Routine etabliert. Derzeit laufen einerseits Studien, in denen getestet wird, unter welchen Umständen man die Behandlung mit TKI absetzen kann, ohne den Patienten einem überhöhten Rezidivrisiko auszusetzen, andererseits wird fieberhaft an neuen Therapieoptionen geforscht, die die Behandlung noch effektiver machen und vor allem den bisher schlecht therapierbaren Patienten mit Philadelphia-Chromosom-positiven Leukämien helfen können.
Die letzten anderthalb Jahrzehnte waren in der Therapie der chronischen myeloischen Leukämie (CML) ganz von den Tyrosinkinaseinhibitoren (TKI) bestimmt. In letzter Zeit gibt es wieder mehr Aufmerksamkeit für das früher gebräuchliche Interferon alpha, weil sich gezeigt hat, dass dessen Wirkmechanismus sich von dem der TKI unterscheidet und dass es die leukämischen Stammzellen im Knochenmark beeinflusst, die gegenüber den TKI weitgehend resistent sind. Es ist also mit einer synergistischen Aktivität zu rechnen, wenn beide Prinzipien kombiniert werden. In der randomisierten Studie SPIRIT konnten Patienten mit einer Kombination aus pegyliertem Interferon alpha 2a und Imatinib tatsächlich signifikant höhere Raten an tiefen molekularen Remissionen erzielen als mit Imatinib alleine [1]. Für Zweitgenerations-TKIs sollte der Effekt noch besser sein, weil sie per se schon höhere Ansprechraten garantieren als Imatinib. Für Nilotinib und Interferon wurden in einer Phase-II-Studie schon vielversprechende Daten berichtet [2], und für Dasatinib wurden in Orlando zwei Phase-II-Studien vorgestellt:
In der skandinavischen NordCML007-Studie erhielten 40 Patienten mit neu diagnostizierter CML pegyliertes Interferon alpha 2b und Dasatinib [3]: Der TKI wurde drei Monate lang als Monotherapie in der Dosierung von 100 mg/d gegeben und dann durch Interferon (drei Monate lang mit 15 µg/Woche und weitere neun Monate lang mit 25 µg/Woche) ergänzt. In einem exploratorischen Vergleich mit der Dasatinib-Monotherapie in der DASISION-Studie schnitt die Kombination deutlich besser aber: Nach zwölf Monaten hatten hier 48% der Patienten eine MR4 (BCR-ABL ≤ 0,01% nach dem Internationalen Standard) erreicht, im DASISION-Kollektiv waren es nur 12% gewesen. Nach 18 Monaten wiesen in der nordischen Studie über 90% der Patienten eine gute molekulare Remission (MMR, d. h. BCR-ABL ≤ 0,1% IS) auf, mit der Dasatinib-Monotherapie waren es etwa 50% gewesen. Die hier verabreichte Kombination war gut verträglich, es gab keine unerwarteten Autoimmunreaktionen und lediglich einen Pleuraerguss vom Grad 2.
Die französische CML-Studiengruppe behandelte in ihrer Phase-II-Studie 61 Patienten mit Dasatinib und pegyliertem Interferon und 18 weitere mit Dasatinib alleine [4]. Nach zwölf Monaten waren hier im Kombinationsarm 70% der Patienten in einer MMR und 30% in einer MR4,5 (BCR-ABL ≤ 0,0032% IS). Es gab allerdings unter der Kombinationstherapie acht schwere Nebenwirkungen.
Die hohen und früh auftretenden Raten an tiefen molekularen Remissionen sind sehr ermutigend: Tiefe Remissionen sind erforderlich, um Patienten künftig ein Absetzen der Therapie mit Aussicht auf ein langes therapie- und rezidivfreies Überleben zu ermöglichen. Es ist also dringend wünschenswert, die Kombination aus TKI und Interferon in randomisierten Studien mit der TKI-Monotherapie zu vergleichen.
Ein neuer Inhibitor mit anderem Mechanismus
ABL001 ist ein potenter Inhibitor von BCR-ABL, der sich von den bisher zugelassenen Substanzen unterscheidet: Er bindet allosterisch in einer Nische der Kinasedomäne von BCR-ABL, die normalerweise vom N-Terminus der ABL1-Kinase eingenommen wird und eine autoregulatorische Funktion ausübt, die aber nach Fusion von ABL mit BCR verlorengeht. ABL001 ahmt diese autoregulatorische Aktivität nach und stellt die negative Regulation der Kinase-Aktivität wieder her. In präklinischen Versuchen konnte die Substanz selektiv das Wachstum BCR-ABL-positiver Zellen hemmen und war auch gegen Mutanten aktiv, die mit einer klinischen Resistenz gegen herkömmliche TKI einhergehen. In Tiermodellen konnte die kombinierte Gabe von ABL001 und Nilotinib Remissionen induzieren, die auch nach Absetzen der Behandlung lange anhielten und in denen keine Resistenz zu beobachten war. In einer Phase-I-Studie wurde ABL001 in steigenden Dosierungen (10–200 mg zweimal täglich) zum ersten Mal beim Menschen eingesetzt, und zwar bei Patienten mit CML, bei denen mindestens zwei Vortherapien mit TKI versagt hatten [6].
In einer Kohorte von zwölf Patienten, die zu Beginn ein hämatologisches Rezidiv gehabt hatten, sprachen alle innerhalb von zwei Monaten mit einer kompletten hämatologischen Remission an. In einer zweiten Kohorte mit einem zytogenetischen Rezidiv zu Beginn war innerhalb von drei bis sechs Monaten unter ABL001 bei zwei Dritteln eine komplette zytogenetische Remission zu sehen, und bei 29 weiteren Patienten, die am Anfang ein molekulares Rezidiv hatten (also nicht in einer guten molekularen Remission gewesen waren), erreichte etwa ein Drittel binnen sechs Monaten eine solche MMR, bei einem Viertel ging die BCR-ABL-Last um mindestens eine, bei weiteren 31% um weniger als eine log-Stufe zurück.
Weil das Ansprechen auf ABL001 offensichtlich dosisabhängig, die maximale tolerierte Dosierung aber noch nicht erreicht ist, wird die Studie fortgeführt, so Oliver Ottmann, Cardiff.
T315I – Ponatinib oder allogene Stammzelltransplantation?
Die "härteste Nuss” in der Therapie der CML und der Philadelphia-Chromosom-positiven ALL sind T315I-Mutationen im BCR-ABL-Fusionsgen. Sie sind resistent gegen nahezu alle zugelassenen Therapien mit Ausnahme des Drittgenerations-TKI Ponatinib. Eine alternative Therapieoption ist – zumindest für jüngere, fitte Patienten mit einem verfügbaren Spender – immer noch eine allogene Stammzelltransplantation. Die wechselseitigen Vorteile dieser beiden Behandlungsmöglichkeiten sind unklar, und weil die T315I-Mutation glücklicherweise recht selten ist, bedurfte es einer internationalen Kollaboration, um wenigstens eine retrospektive Vergleichsstudie auf den Weg zu bringen [7]. In sie gingen 184 Patienten ein; 128 davon waren in der Phase-II-Studie PACE [8] mit Ponatinib behandelt worden, während die Daten der übrigen 56 allogen transplantierten Patienten aus dem Register der European Group for Bone and Marrow Transplantation (EBMT) extrahiert wurden.
90 Patienten waren vor Beginn der jeweiligen Therapie in der chronischen Phase einer CML gewesen, 26 in der akzelerierten Phase, 41 in der Blastenkrise, die restlichen 27 hatten an einer Ph-positiven ALL gelitten. Die mit Ponatinib behandelten Patienten waren zu Beginn der Intervention signifikant älter als die transplantierten (53 vs. 45 Jahre; p = 0,006). Primärer Endpunkt des Vergleichs war das Gesamtüberleben, und hier schnitt Ponatinib bei CML in der Kohorte in chronischer Phase signifikant besser ab, in der Blastenkrise hingegen signifikant schlechter. Bei den CML-Patienten in der akzelerierten Phase war kein Unterschied zu erkennen, während die Patienten mit Ph-positiver ALL von der Transplantation deutlich, wenngleich nicht signifikant stärker profitierten (Tab. 1).
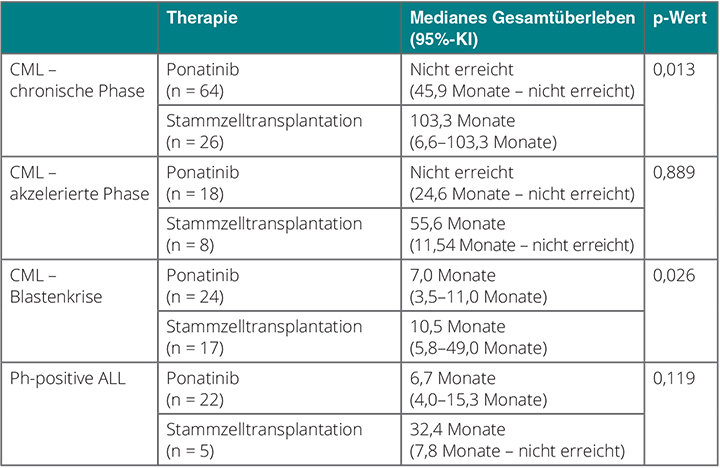
Die allogene Stammzelltransplantation bleibt damit weiter eine valide und potenziell kurative Therapieoption zumindest für CML-Patienten mit T315I-Mutation im BCR-ABL-Gen, die sich bereits in der Blastenkrise befinden. Auch bei Ph-positiver ALL mit dieser Anomalie scheint sie Vorteile zu bieten, wenngleich der Unterschied statistisch nicht zu belegen ist. Bei Patienten in der chronischen Phase einer CML scheint hingegen die medikamentöse Therapie mit dem Drittgenerations-TKI Ponatinib Vorteile zu haben – auch wenn man einschränken muss, dass dieser Vergleich nicht randomisiert und die Nachbeobachtungszeit mit bis zu vier Jahren nicht sehr lang war, so Franck Nicolini, Pierre-Benite.
STIM-1: auch nach über fünf Jahren vier von zehn Patienten rezidivfrei
In der einarmigen französischen STIM-1-Studie (STop IMatinib) konnte erstmals gezeigt werden, dass das Absetzen eines TKI nach mindestens zwei Jahren mit nicht messbarer minimaler Resterkrankung sicher ist. In der Langzeit-Nachbeobachtung median 65 Monate nach Absetzen von Imatinib, die Gabriel Etienne, Bordeaux, in Orlando präsentierte, bestätigte sich, dass das rezidivfreie Überleben auf einem Niveau von rund 40% auch nach einer so langen Zeit anhält [9]. Die überwiegende Mehrzahl der molekularen Rezidive trat innerhalb der ersten sechs Monate nach Absetzen der Behandlung auf und reagierte prompt auf die erneute Behandlung mit einem TKI.
Für die Patienten, die die ersten sechs Monate ohne Rezidiv bleiben, sinkt das Risiko, bis zu zwei Jahre nach Absetzen eines zu erleiden, auf 10%. Nach mehr als 24 Monaten ist bislang kein einziges molekulares Rezidiv aufgetreten. In einer multivariaten Analyse erwies sich der Sokal-Risiko-Score als prädiktiv: Weniger als die Hälfte der Patienten mit niedrigem Sokal-Risiko erlitt ein Rezidiv, bei solchen mit hohem Sokal-Score lag das Risiko dagegen bereits nach sechs Monaten bei etwa 80%. Wirklich beruhigend ist vor allem, dass nach einem Rezidiv durch Wiederaufnahme der TKI-Therapie bei 55 von 57 Patienten die BCR-ABL-Titer wieder unter die Nachweisgrenze gedrückt werden konnten.
Immuncheckpoint-Inhibitoren auch bei CML?
Die leukämischen Stammzellen bei der CML sind unempfindlich gegen BCR-ABL-TKI und können dadurch nicht eliminiert werden. Unklar ist, warum dennoch ein Teil der Patienten die TKI-Therapie beenden kann, ohne ein Rezidiv zu erleiden. Vermutlich spielen immunologische Mechanismen eine Rolle, die die leukämischen Stammzellen in Schach halten. In der EURO-SKI-Studie, in der ein Absetzen von TKI geprobt wird, wurde versucht, diesen Mechanismen auf die Spur zu kommen:
Andreas Burchert, Marburg, fokussierte dabei auf plasmazytoide dendritische Zellen, die als wichtige Regulatoren von Immunantworten gelten [10]. Werden sie aktiviert, so regulieren sie bestimmte Marker-Antigene hoch wie MHC-Antigene der Klasse II und CD86 (auch als B7.2 bekannt). CD86 spielt unter anderem eine Rolle bei inhibitorischen Immun-Checkpoint-Mechanismen, indem es an den Rezeptor CLTA-4 auf T-Lymphozyten bindet.
Die bei 123 Patienten aus der EURO-SKI-Studie und an Kontrollprobanden per Durchflusszytometrie bestimmten Daten belegten erstmals, dass die Anwesenheit von mehr als 30% CD86-positiven plasmazytoiden dendritischen Zellen im peripheren Blut hochgradig mit dem Rezidivrisiko nach Absetzen assoziiert ist (p = 0,0025). Noch schärfer ist die Trennung von Hoch- und Niedrigrisiko-Gruppen, wenn man einen Anteil von mehr oder weniger als 95 CD86-positiven Zellen an 10.000 Lymphozyten als Cut-off-Wert verwendet (p < 0,0001).
Die langfristige Anwesenheit von CD86-positiven plasmazytoiden dendritischen Zellen könnte also durch den über die Interaktion von CD86 und CTLA-4 vermittelten inhibitorischen Effekt auf T-Lymphozyten zur Erschöpfung der T-Zell-Reserve führen und dadurch eine Immunreaktion gegen die CML-Stammzellen konterkarieren – was wiederum zum Rezidiv nach Absetzen von TKI führen könnte. Sollten sich diese Befunde bestätigen, so könnte sich dadurch eine neue therapeutische Option eröffnen: Der Anti-CTLA-4-Antikörper Ipilimumab könnte möglicherweise auch bei der CML ein molekulares Rezidiv nach Absetzen von TKI verhindern und die Persistenz leukämischer Stammzellen im Knochenmark durchbrechen.
Literatur
1. Preudhomme C et al. Imatinib plus peginterferon alfa-2a in chronic myeloid leukemia. N Engl J Med 2010; 363: 2511-21.
2. Nicolini FE et al. Nilotinib and peginterferon alfa-2a for newly diagnosed chronic-phase chronic myeloid leukaemia (NiloPeg): a multicentre, non-randomised, open-label phase 2 study. Lancet Haematology 2015; 2: e37-46.
3. Hjorth-Hansen H et al. ASH 2015, Abstract #477.
4. Roy L et al. ASH 2015, Abstract #134.
5. Giles FJ et al. ASH 2015, Abstract #479.
6. Ottmann O et al. ASH 2015, Abstract #138.
7. Nicolini FE et al. ASH 2015, Abstract #480.
8. Cortes JE et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med 2013; 369: 1783-96.
9. Etienne G et al. ASH 2015, Abstract #345.
10. Burchert A et al. ASH 2015, Abstract #599.

Josef Gulden