Die Klassifikation von Hirntumoren basierte in den letzten hundert Jahren vor allem auf Konzepten zur Histogenese, d. h. auf der Vorstellung, dass die Tumoren anhand ihrer mikroskopisch erkennbaren Ähnlichkeiten mit unterschiedlichen Ursprungszellen und ihres wahrscheinlichen Differenzierungsgrades klassifizieren kann. Definiert wurden solche histologischen Ähnlichkeiten überwiegend aufgrund morphologischer Kriterien, die mittels H & E-Färbungen im Lichtmikroskop, der immunhistochemisch nachgewiesenen Expression gewebespezifischer Proteine und der elektronenmikroskopischen Charakterisierung gewonnen wurden. So arbeitete die letzte WHO-Klassifikation von 2007 noch mit einer strikten Trennung nach astrozytärem und oligodendroglialem Phänotyp, ohne zu berücksichtigen, ob die verschiedenen astrozytären Tumoren sich klinisch ähnlich oder unterschiedlich verhielten [3].
Die vergangenen beiden Jahrzehnte brachten für die häufigen und auch für einige der selteneren Entitäten die Aufklärung der genetischen Grundlagen der Tumorentstehung, mit denen eine „natürlichere“ Klassifikation dieser Tumoren möglich sein sollte. Einige dieser kanonischen genetischen Veränderungen waren 2007 bereits bekannt, aber zu dieser Zeit sah man sich noch nicht imstande, damit strikt spezifische Entitäten zu definieren. Die betreffenden Mutationen wurden lediglich als prognostische oder prädiktive Marker gesehen, die sich den durch die konventionelle Histologie etablierten klassischen Diagnosekategorien unterzuordnen hatten.
Bahnbrechend für die Neuausrichtung war eine 2014 von der International Society of Neuropathology organisierte Konferenz in Haarlem, die Richtlinien für die Inkorporierung der molekularen Befunde in die Diagnose von Hirntumoren etablierte und damit die Voraussetzungen für die jetzt publizierte Revision der Klassifikation von 2007 schuf [4]. Diese führt nach hundert Jahren zahlreiche molekulare Parameter in die Klassifikation von Hirntumorentitäten ein [1]. Getragen wurde die Arbeit durch ein internationales Konsortium von 117 Spezialisten aus 20 Ländern; kontroverse Fragestellungen wurden bei einer dreitägigen Konsensuskonferenz mit 35 Neuropathologen, Neuroonkologen und Naturwissenschaftlern aus zehn Ländern diskutiert.
Diffuse Gliome
Die nosologische Verschiebung hin zu einer Klassifikation, die Phäno- und Genotyp gleichermaßen berücksichtigt, zeigt sich beispielhaft in der Klassifikation der diffusen Gliome (Abb. 1): Vor allem werden nun im Gegensatz zur alten Klassifikation alle diffus infiltrierenden Gliome zusammengefasst, egal ob astrozytär oder oligodendroglial – und zwar nicht alleine nach Wachstumsmuster und Verhaltens, sondern insbesondere auf der Grundlage gemeinsamer Treibermutationen in den Genen für die Isocitrat-Dehydrogenase 1 und 2 (IDH1 und IDH2). Diese Klassifikation bildet die Dynamik der Pathogenese auf der molekularen und morphologischen Ebene ab. Für die Prognosestellung hat sie den Vorteil, dass Tumoren mit den gleichen prognostischen Markern zusammengruppiert werden. Aus der Sicht von behandelndem Arzt und Patient kann sie die Therapie (mit konventionellen oder zielgerichteten Ansätzen) für biologisch und genetisch ähnliche Entitäten steuern.
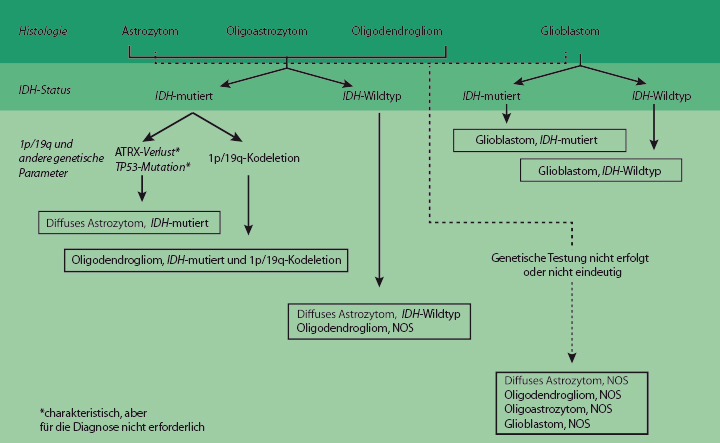
Die diffusen Gliome umfassen in der neuen Klassifikation astrozytäre Tumoren und Oligodendrogliome des WHO-Grads II und III, die Glioblastome des Grads IV und die damit verwandten diffusen Gliome des Kindesalters (s. weiter unten). Von den diffusen Gliomen abgetrennt werden durch dieses Vorgehen Astrozytome mit einem schärfer umrissenen Wachstumsmuster, die keine Veränderungen in den IDH-Genen, dafür aber häufig BRAF-Mutationen (pilozytische Astrozytome, pleomorphe Xanthoastrozytome) oder TSC1/2-Mutationen aufweisen (subependymale Riesenzell-Astrozytome). Hiermit wurden sozusagen die Stammbäume der Tumoren neu gezeichnet: So sind zum Beispiel diffuse Astrozytome und Oligodendrogliome nun stärker miteinander verwandt als diffuse und pilozytische Astrozytome.
Diffuses und anaplastisches Astrozytom
Die diffusen Astrozytome vom WHO-Grad II und die anaplastischen Astrozytome vom Grad III werden nun jeweils in die Kategorien IDH-mutiert, IDH-unmutiert und NOS (not otherwise specified) unterteilt. Bei Grad-II- ebenso wie bei Grad-III-Tumoren fällt die überwiegende Mehrzahl in die Kategorie IDH-mutiert, sofern ein IDH-Test verfügbar ist. Als IDH-Wildtyp gelten Tumoren, die negativ für Mutationen in Kodon 132 von IDH1 und Kodon 172 von IDH2 sind und, soweit verfügbar, auch auf Proteinebene immunhistochemisch kein R132H-mutiertes IDH1 aufweisen. Man sollte jedoch beachten, dass solche IDH-Wildtyp-Astrozytome selten sind; daher ist beim diffusen Astrozytom ein Tumor niedrigeren Grads wie zum Beispiel ein Gangliogliom auszuschließen, und beim anaplastischen Astrozytom mit IDH-Wildtyp finden sich meist genetische Veränderungen, die charakteristisch für ein Glioblastom mit IDH-Wildtyp sind. Ist keine vollständige IDH-Mutationstestung verfügbar, so würde die Diagnose diffuses oder anaplastisches Astrozytom, NOS, lauten.
Die Angabe des WHO-Grads wird bis auf Weiteres empfohlen: Es gibt zwar Hinweise darauf, dass das Vorliegen einer IDH-Mutation größere prognostische Bedeutung hat als der WHO-Grad, aber die Befunde dazu sind teilweise widersprüchlich.
Aus der Klassifikation gestrichen wurden das protoplasmatische und das fibrilläre Astrozytom, sodass als Variante der diffusen Astrozytome, IDH-mutiert, lediglich das gemistozytische Astrozytom übrigbleibt. Als eigene Entität ebenfalls gestrichen wurde die Gliomatosis cerebri, die heute eher als ein spezielles, bei vielen Gliomen auftretendes Wachstumsmuster angesehen wird, dessen biologische Ursachen noch unklar sind.
Glioblastome
Bei den Glioblastomen werden in der neuen Klassifikation drei Untertypen unterschieden:
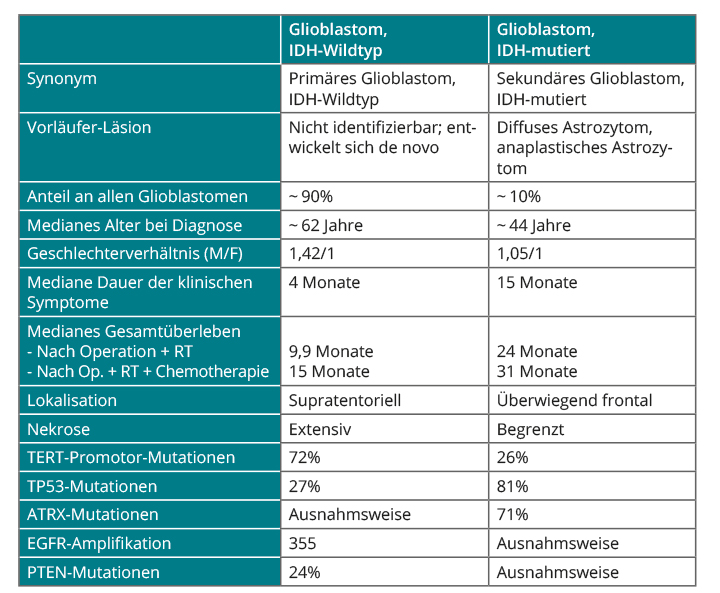
1. Das „Glioblastom, IDH-Wildtyp“, macht ungefähr 90% aller Fälle aus, entspricht meist dem klinisch definierten primären Glioblastom und findet sich überwiegend bei über 55-Jährigen.
2. Das „Glioblastom, IDH-mutiert“ stellt etwa 10% aller Fälle und entspricht dem sogenannten sekundären Glioblastom, das sich aus einem diffusen Gliom niedrigeren Grades entwickelt hat und vor allem junge Patienten befällt.
3. Die Diagnose „Glioblastom, NOS“ wird gestellt, wenn eine vollständige IDH-Mutationsanalyse nicht möglich ist. Dabei ist zu beachten, dass bei jüngeren Patienten mit negativer Immunhistochemie bezüglich R132H IDH1 eine Sequenzierung empfohlen wird (übrigens auch bei diffusen Grad-II- und Grad-III-Gliomen), während solche Mutationen bei über 55-Jährigen kaum vorkommen, sodass eine Sequenzierung im Fall der negativen Immunhistochemie nicht als erforderlich angesehen wird.
Als provisorische neue Entität wurde das epitheloide Glioblastom eingeführt, das zusammen mit dem Riesenzell-Glioblastom und dem Gliosarkom zu den IDH-Wildtyp-Glioblastomen gezählt wird. Es tritt vorzugsweise bei Kindern und jungen Erwachsenen auf und weist oft eine BRAF-V600E-Mutation auf, die sich immunhistochemisch nachweisen lässt.
Als neues Muster wurde das Glioblastom mit primitiver neuronaler Komponente eingeführt, früher als Glioblastom mit PNET-artiger Komponente bezeichnet. Es imponiert zwar als diffuses Astrozytom (oder seltener Oligodendrogliom), enthält aber primitive Zellen mit neuronaler Differenzierung und zeigt eine Tendenz zur Disseminierung über den Liquor. Wird dieses Muster erkannt, so sollte man deshalb eine Tumoraussaat entlang der kraniospinalen Achse ausschließen.
Oligodendrogliome
Die Diagnose eines Oligodendroglioms oder eines anaplastischen Oligodendroglioms erfordert den Nachweis einer IDH-Mutation sowie einer 1p/19q-Kodeletion. Fällt die Immunhistochemie bezüglich R132H IDH1 negativ aus, so wird die Sequenzierung der relevanten IDH1- und IDH2-Kodons empfohlen. Ist das nicht möglich oder sind die Ergebnisse nicht eindeutig, so wird ein Oligodendrogliom, NOS, diagnostiziert. Bei einem solchen anaplastischen Oligodendrogliom kann auch nach genetischen Merkmalen eines Glioblastoms gesucht werden.
Es gibt Tumoren des Kindesalters, die histologisch wie ein Oligodendrogliom aussehen, aber nicht die genannten genetischen Alterationen aufweisen. Sie sollten bis auf Weiteres als „Oligodendrogliom, NOS“, diagnostiziert werden, sofern seltenere, histologisch ähnliche Formen wie ein pilozytisches Astrozytom, ein dysembryoblastischer neuroepithelialer Tumor oder ein klarzelliges Ependymom ausgeschlossen sind.
Oligoastrozytome
Die Diagnose eines Oligoastrozytoms wird in der neuen Klassifikation nicht mehr empfohlen. Man kann bei diesen Tumoren, die histologisch beide Komponenten enthalten, fast immer mithilfe genetischer Tests entweder ein Astrozytom oder ein Oligodendrogliom diagnostizieren. Grad-II- und Grad-III-Oligoastrozytome sind daher als NOS zu kennzeichnen als Hinweis darauf, dass die molekulare Testung nicht möglich ist. Das gilt bis auf Weiteres auch für die seltenen Fälle, in denen tatsächlich phänotypisch und genotypisch räumlich getrennte oligodendrogliale und astrozytäre Komponenten nachzuweisen sind.
Pädiatrische diffuse Gliome
Der Nachweis spezifischer genetischer Aberrationen in pädiatrischen diffusen Gliomen gestattet mittlerweile, einige Entitäten von den entsprechenden Tumoren bei Erwachsenen abzugrenzen. Ein gut zu definierender Typ von Tumoren, der vor allem bei Kindern (nur gelegentlich auch bei Erwachsenen) auftritt, ist charakterisiert durch die K27M-Mutation im H3F3A-Gen für das Histon H3 oder seltener im eng verwandten Gen HIST1H3B, ein diffuses Wachstumsmuster und eine Lokalisation nahe der Mittellinie (z. B. Thalamus, Hirnstamm, Rückenmark). Diese neue Entität wird künftig als diffuses Mittellinien-Gliom, H3 K27M-mutiert, geführt und umfasst auch das frühere diffuse intrinsische pontine Gliom (DIPG). Die spezifischen molekularen Veränderungen könnten als Ansatzpunkte für die Entwicklung von Therapien dienen.
Andere Astrozytome
Das früher so genannte pleomorphe Xanthoastrozytom mit anaplastischen Eigenschaften wird in der Aktualisierung der Klassifikation als eigene Entität mit dem Namen anaplastisches pleomorphes Xanthoastrozytom, WHO-Grad III, geführt. Ein Grading als anaplastisch erfordert den Nachweis von mindestens fünf Mitosen pro zehn HPF (High-power fields), eine Nekrose kann vorhanden sein, aber ihre Bedeutung bei fehlender erhöhter mitotischer Aktivität ist unklar. Die Überlebenszeiten dieser Patienten sind kürzer als bei pleomorphen Grad-II-Xanthoastrozytomen.
Das Grading von pilomyxoiden Astrozytomen wurde in der neuen Klassifikation sozusagen ausgesetzt, weil unklar ist, ob damit automatisch ein WHO-Grad II einhergeht.
Ependymome
Das Grading der Ependymome wird infrage gestellt, weil es schwierig zu verifizieren und in seinem klinischen Nutzen fragwürdig ist. Eine differenziertere molekulare Charakterisierung dürfte hier Fortschritte bringen, ein genetisch definierter Subtyp ist bereits akzeptiert: das Ependymom mit RELA-Fusion, das die Mehrzahl aller supratentoriellen Tumoren bei Kindern ausmacht. Ob der immunhistochemische Nachweis von L1CAM einen validen Surrogatmarker dafür darstellt, ist noch unklar. Gestrichen wurde aus der Klassifikation das zelluläre Ependymom, weil es stark mit Standard-Ependymomen überlappt.
Neuronale und gemischt neuronale-gliale Tumoren
Die neue Entität des diffusen leptomeningeal-glioneuronalen Tumors war bisher unter verschiedenen Namen bekannt, v. a. als disseminierter Oligodendroglia-artiger leptomeningealer Tumor des Kindesalters. Er imponiert als diffuse leptomeningeale Erkrankung mit oder ohne erkennbare Parenchym-Komponente, meist im Rückenmark von Kindern und zeigt eine monomorphe, klarzellige Glia-Morphologie, die an ein Oligodendrogliom erinnert. Häufig wird aber zusätzlich zu OLIG2 und S-100 auch Synaptophysin exprimiert. Manchmal findet sich eine neuronale Komponente, und die Tumoren enthalten meist BRAF-Fusionen sowie eine 1p-Deletion mit oder ohne gleichzeitige 19q-Deletion. IDH-Mutationen findet man aber nicht. Die nosologische Position dieser Tumoren ist derzeit unklar, einige pathologische und genetische Eigenschaften deuten auf eine Verwandtschaft mit pilozytischen Astrozytomen oder glioneuronalen Tumoren hin. Die Prognose der Patienten ist variabel, die Tumoren wachsen zwar langsam, aber es kann zu einem sekundären Hydrozephalus kommen.
Unklar ist die nosologische Position eines multinodulären und vakuolisierten Musters, das eventuell eine Verwandtschaft zu Ganglionzell-Tumoren aufweist, möglicherweise wenig aggressiv ist und eher eine Fehlbildung darstellt.
Medulloblastome
Das Gliederungskonzept der Medulloblastome wurde grundlegend überarbeitet; Neuerungen ergeben sich v. a. aus der Kombination histologischer und molekularer Klassifikationsmuster. Es gibt einerseits lange etablierte und klinisch brauchbare histologische Varianten (z. B. desmoplastisch/nodulär, Medulloblastom mit ausgeprägter Nodularität, großzellige und anaplastische Medulloblastome), andererseits sind mittlerweile vier genetisch definierte Kategorien allgemein akzeptiert: Medulloblastome mit Aktivierung des Wnt- bzw. des Sonic Hedgehog (SHH)-Signalwegs sowie die „Gruppe 3“ und „Gruppe 4“, in denen diese Signalwege genetisch nicht verändert sind. Diese histologische und genetische Differenzierung geht teilweise mit Unterschieden in Prognose und therapeutischen Konsequenzen einher. Die Klassifikation beschränkt sich darauf, „genetisch definierte“ und „histologisch definierte“ Varianten aufzulisten – in der Erwartung, dass ein Pathologe, der die molekulare Klassifikation durchführen kann, eine integrierte Diagnose unter Angabe der molekularen Gruppe und des histologischen Phänotyps stellen wird. Eine Anzahl der klinisch besonders relevanten Diagnosen zeigt Tab. 1.
Dieser modulare und integrierte Ansatz ist neuartig, so die Autoren der Klassifikation, und wird für zukunftweisend gehalten, weil man nun auch das stetig zunehmende Wissen über Tumorgenetik und Phänotyp-Genotyp-Korrelation leichter und zeitnäher integrieren kann.
Andere embryonale Tumoren
Auch die Klassifikation der embryonalen Nicht-Medulloblastom-Tumoren hat sich erheblich gewandelt. So wurde der Terminus primitiver neuroektodermaler Tumor (PNET) gestrichen, weil sich bei vielen dieser seltenen Tumoren eine Amplifikation der C19MC-Region auf Chromosom 19 (19q13.42) findet. Sie werden jetzt unter dem Begriff embryonaler Tumor mit mehrschichtigen Rosetten (embryonal tumor with multilayered rosettes, ETMR) mit C19MC-Veränderung zusammengefasst und enthalten beispielsweise die früheren Entitäten embryonale Neuropil-reiche Tumoren mit echten Rosetten (ETANTR), Ependymoblastom sowie Medulloepitheliom. Bei Fehlen einer C19MC-Amplifikation sollte ein histologisch als ETANTR/ETMR imponierender Tumor als embryonaler Tumor mit mehrschichtigen Rosetten, NOS, diagnostiziert werden. Bei den Medulloepitheliomen gibt es offenbar einige, die weiterhin so zu bezeichnen sind, weil sie keine C19MC-Amplifikation aufweisen.
Atypische teratoide/rhabdoide Tumoren (AT/RT) sind nun definiert durch Veränderungen im INI1-Gen oder sehr selten im BRG1-Gen, die sich immunhistochemisch als fehlende nukleäre Expression der entsprechenden Proteine nachweisen lassen. Ein Tumor mit dieser Histologie, aber ohne die entsprechenden molekularen Defekte, wird künftig rein deskriptiv als embryonaler ZNS-Tumor mit rhabdoiden Eigenschaften geführt.
Für die übrigen embryonalen Tumoren wurde in der neuen Klassifikation eine Sammelkategorie embryonale ZNS-Tumoren, NOS, eingerichtet – in der Erwartung, dass das Auftauchen neuer molekularer Marker hier zu einer weiteren Differenzierung führen wird.
Nervenscheidentumoren
Bei der Klassifikation der kraniellen und paraspinalen Nervenscheidentumoren hat sich nicht viel geändert. Das melanotische Schwannom wurde als neue Entität abgegrenzt, weil man es klinisch und genetisch (z. B. Assoziation mit dem Carney-Komplex und dem PRKAR1A-Gen) eindeutig vom konventionellen Schwannom differenzieren kann. Die hybriden Nervenscheidentumoren wurden aufgenommen, weil sie zunehmend häufiger in einer Reihe von Kombinationen diagnostiziert werden, auch wenn sie möglicherweise nicht einen einzigen Subtyp darstellen. Schließlich führt die Klassifikation nun zwei Subtypen maligner peripherer Nervenscheidentumoren (MPNST) auf, die wegen ihrer klinischen Charakteristika als eigene Varianten gelten können: den epitheloiden MPNST und den MPNST mit perineuraler Differenzierung.
Meningeome
An der Klassifikation der Meningeoma hat sich lediglich ein Aspekt geändert: Die Invasion ins Gehirn wurde als Kriterium für die Diagnose eines atypischen Meningeoms vom WHO-Grad II festgeschrieben und tritt damit als histologischer Marker gleichberechtigt an die Seite einer Zahl von mindestens vier Mitosen.
Solitärer fibröser Tumor/Hämangioperizytom
In den letzten zehn Jahren ist immer klarer geworden, dass solitäre fibröse Tumoren und Hämangioperizytome überlappende, wenn nicht gar identische Entitäten darstellen, weil sie beide Inversionen am Locus 12q13 mit der Folge einer Fusion des NAB2- und des STAT6-Gens aufweisen. Die daraus resultierende nukleäre Expression des STAT6-Proteins lässt sich immunhistochemisch nachweisen. Die Bezeichnung solitärer fibröser Tumor/Hämangioperizytom ist etwas sperrig und könnte sehr wohl in der nächsten Ausgabe der Klassifikation durch einen einfacheren Terminus ersetzt werden, so die Autoren.
Entgegen den bisherigen Gewohnheiten bei der Klassifikation von ZNS-Tumoren wurde außerdem entschieden, unterschiedliche Malignitätsgrade dieser Tumoren nicht durch separate Namen zu kennzeichnen, sondern das Grading innerhalb des Begriffs solitärer fibröser Tumor/Hämangioperizytom zu implementieren: Ein Grad-I-Tumor entspricht also hier nun in der Regel dem bisherigen solitären fibrösen Tumor mit hohem Kollagenanteil, relativ geringer Zellularität und Spindelzellen. Als Grad-II-Tumor wird das bisherige Hämangioperizytom mit höherer Zellularität, weniger Kollagen, prallen Zellen und „staghorn“-Sinusoiden bezeichnet, und Grad-III-Tumoren entsprechen der alten Einheit des anaplastischen Hämangioperizytoms mit mindestens fünf Mitosen. Die Autoren versprechen sich von diesem für ZNS-Tumoren neuen Einteilungsmodus eine größere Flexibilität bei der künftigen Implementierung neuer molekularer Marker in das Grading dieser Tumoren.
Lymphome und histiozytäre Tumoren
Bei diesen Tumoren wurden in der aktualisierten Version der Klassifikation alle Änderungen der Einteilung übernommen, die sich in den vergangenen zehn Jahren bei systemischen Lymphomen und histiozytären Neoplasien ergeben haben.
Fazit
Der wichtigste, zukunftweisende Aspekt der neuen WHO-Klassifikation für ZNS-Tumoren von 2016 ist die erstmalige Integration molekularer Parameter in die Diagnostik mit weitreichenden Auswirkungen nicht nur auf die Nomenklatur und die Nosologie, sondern auch auf die Struktur des pathologischen Berichts. Wahrscheinlich ist damit allerdings erst ein Zwischenstadium in einem Prozess erreicht, der in künftigen Versionen der Klassifikation noch zu sehr viel weitergehenden Veränderungen führen wird. Das ist nicht l´art pour l´art: Vielmehr erhofft man sich durch die verstärkt molekulare Ausrichtung eine objektivere und präzisere Definition der einzelnen Entitäten mit dem Ziel, dem jeweiligen Patienten eine immer individualisiertere Therapie anbieten zu können.
Abgesehen davon wird die neue Klassifikation in künftigen klinischen Studien zu einer besseren Stratifizierung der Patienten und damit zu präziseren Ergebnissen führen. Auch wenn es noch Situationen gibt, in denen auch ohne Vorliegen molekularer Daten eine Diagnose gestellt werden muss, so sind diese Gruppen jetzt doch deutlich klarer definiert und lassen sich besser von den molekular definierten Entitäten absetzen. Letztlich ist zu hoffen, so die Autoren, dass die neue Klassifikation die klinische, experimentelle und epidemiologische Forschung zum Wohl der Patienten mit Hirntumoren fördern wird.
Josef Gulden
Literatur
1. Louis DN et al. World Health Organization Histological Classification of Tumours of the Central Nervous System. International Agency for Research on Cancer, France, 2016.
2. Louis DN et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol 2016; 131: 803-20.
3. Louis DN et al. World Health Organization histological classification of tumours of the central nervous system. International Agency for Research on Cancer, Lyon, 2007.
4. Louis DN et al. International Society of Neuropathology-Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol 2014; 24: 429-35.