Polycythaemia vera: Pathogenese, Diagnostik und Therapie
Susanne Isfort, Tim H. Brümmendorf, Steffen Koschmieder
Definition der Polycythaemia vera
Die Polycythaemia vera (PV) gehört zu den chronischen myeloproliferativen Neoplasien (MPN). Sie zeichnet sich meist durch eine Hyperplasie aller drei Zellreihen (Leukozyten, Erythrozyten sowie Thrombozyten) aus. Die Variante mit ausschließlicher Vermehrung der roten Zellreihe wird Polycythaemia vera rubra genannt, sie ist jedoch äußerst selten. Charakteristisch für diese Formen der PV ist der erhöhte Hämoglobin- und/oder Hämatokrit-Wert. Darüber hinaus wurde kürzlich eine sogenannte „maskierte PV“ beschrieben, die mit niedrigeren Hämoglobin-/Hämatokrit-Werten einhergeht [1].
Die PV ist eine seltene Erkrankung mit einer Inzidenz von ungefähr 2/100.000 Einwohner pro Jahr [2]. Das mediane Erkrankungsalter liegt zwischen 60 und 65 Jahren. Die PV verläuft klinisch in zwei Phasen: Einer chronischen hyperplastischen Phase, welche durch eine Überproduktion von Blutzellen, vor allem Erythrozyten, gekennzeichnet ist, folgt die sogenannte Spätphase, in es zu einem Übergang der Erkrankung in eine sekundäre Myelofibrose kommt, welche durch extramedulläre Blutbildung mit konsekutiver Splenomegalie sowie durch Zytopenien charakterisiert ist. Bei einem kleineren Teil der Patienten kommt es zu einem Übergang in eine sekundäre akute Leukämie, die eine infauste Prognose hat und in den meisten Fällen tödlich endet.
Pathogenese
Der genaue Pathomechanismus der PV ist noch nicht bekannt. Allerdings konnte bei über 95% der Patienten eine Mutation im Exon 14 des Gens für die Janus-Kinase 2 (JAK2) nachgewiesen werden. Das entstehende Protein JAK2V617F verfügt über eine gesteigerte Tyrosinkinase-Aktivität. Die Janus-Kinase 2 ist bei Gesunden essenziell für die Blutbildung. Über sie führen Zytokine wie Erythropoetin, Thrombopoetin oder G-CSF zu einer Aktivierung der STAT-Moleküle in der Zelle und zu einer Proliferation hämatopoetischer Zellen [3]. Das mutierte JAK2V617F-Protein führt zu einer Wachstumsfaktor-unabhängigen Aktivierung dieser Signale [4]. Hierdurch kommt es zu einer unkontrollierten Proliferation von Blutzellen. In seltenen Fällen liegt der JAK2-Aktivierung auch eine Mutation im Exon12 des JAK2-Gens zugrunde.
Neue genetische und molekulargenetische Untersuchungen haben in den letzten Jahren unser Verständnis der Pathogenese der PV sowie der teilweise unterschiedlichen klinischen Verläufe vertieft. So konnten Spivak et al. anhand eines genetischen Profils mittels Oliginukleotid-Microarray-Analysen zwei unterschiedliche Verlaufsformen der PV definieren [5]. Es fanden sich 102 Gene, welche bei männlichen und weiblichen Patienten mit PV gleichverteilt waren. Durch diese 102 Gene konnten die insgesamt 19 Patienten in zwei unterschiedliche Gruppen aufgeteilt werden. Diese unterschieden sich nicht, was die Altersverteilung, die JAK2-Allellast oder Blutwerte (Leukozyten, Thrombozyten) anbelangt, eine Gruppe zeichnete sich jedoch durch einen signifikant aggressiveren Verlauf aus u. a. im Sinne einer kürzeren Zeit bis zur Progression, einer höheren Rate an thromboembolischen Ereignissen und einer erhöhten leukämischen Transformationsrate.
Ortmann et al. [6] veröffentlichten 2014 eine Analyse von Patienten mit PV, welche mindestens zwei verschiedene Mutationen (JAK2V617F und TET2) aufwiesen. Dabei stellte sich heraus, dass die Reihenfolge, in welcher diese Mutationen erworben werden, einen erheblichen Einfluss auf den klinischen Verlauf des Patienten hat. Patienten, welche erst eine TET2-Mutation und dann im weiteren Verlauf eine JAK2-Mutation erwarben, hatten einen indolenteren Verlauf Ihrer Erkrankung. Patienten mit einer initialen JAK2-Mutation wiesen hingegen ein erhöhtes Thromboserisiko auf.
Beide Ansätze zeigen, dass die molekulare Diagnostik dieser Erkrankung einen immer höheren Stellenwert einnimmt und uns eventuell in einigen Jahren erlauben wird, schon frühzeitig Schlüsse über die Prognose zu ziehen.
Klinisches Bild
Die meisten Patienten mit PV weisen Symptome auf. Allerdings sind diese oft unspezifisch und führen erst spät zur korrekten Diagnose. Das häufigste Symptom ist die Fatigue, welche in über 90% der Fälle vorliegt [7]. Des Weiteren leiden die Patienten an Kopfschmerzen und anderen Symptomen, welche am ehesten durch Mikrozirkulationsstörungen verursacht werden. Ein PV-typisches, nahezu pathognomonisches Symptom ist die Erythromelalgie, eine Durchblutungsstörung der Hände und Füße, welche sich durch Brennen und Rötung äußert und sehr gut auf die Therapie mit Azetylsalizylsäure (ASS) anspricht. Außerdem können die typischen B-Symptome (Fieber, Nachtschweiß, Gewichtsverlust) auftreten.
Der aquagene Pruritus stellt ein weiteres typisches Symptom der PV dar. Diese Patienten leiden unter starkem Juckreiz, v. a. nach Berührung der Haut mit Wasser [8]. Oftmals ist der aquagene Pruritus ein initialer Hinweis auf eine PV, und zuweilen wird die Diagnose des Symptoms Jahre vor der Diagnose einer PV gestellt.
Die wichtigsten Komplikationen der PV sind Thrombosen bzw. thromboembolische Ereignisse. Diese treten mit einer Häufigkeit von 3–5% pro Jahr nach Diagnose auf [9]. Die Behandlung der PV zielt in erster Linie auf eine Vermeidung solch thromboembolischer Ereignisse ab. Blutungskomplikationen spielen bei der PV eine eher untergeordnete Rolle.
Diagnostik
Die Diagnostik einer Polycythaemia vera umfasst in erster Linie Laborparameter. Im Blutbild zeigt sich typischerweise eine führende Erythrozytose, meist mit Leukozytose und Thrombozytose, im Differenzialblutbild jedoch keine Vermehrung von Blasten. Der Erythropoetin-Spiegel ist typischerweise erniedrigt (DD zur sekundären Polyglobulie). CRP und/oder Ferritin sollten zum Ausschluss eines reaktiven Geschehens bestimmt werden. Die aktuell gültigen Diagnosekriterien nach der WHO-Klassifikation von 2008 sind in Tabelle 1 zu finden [10].Der Nachweis einer Exon 14 (V617F)-Mutation der Janus-Kinase 2 gelingt in über 95% aller Patienten, andere Mutationen, welche sonst bei den verwandten MPN wie essenzielle Thrombozythämie (ET) oder primäre Myelofibrose (PMF) nachweisbar sind (Mutationen im Calreticulin-Gen, MPL-Mutationen), sind bei der Polycythaemia vera gar nicht oder nur in Einzelfällen beschrieben. Bei Negativität für eine klassische V617F-Mutation lohnt sich die Bestimmung einer Exon 12-Mutation, welche in seltenen Fällen vorliegen kann. Außerdem kann in Einzelfällen die Bestimmung einer SH2B3 (LNK)-Mutation den Nachweis eines klonalen Markers im Sinne des zweiten Major-Kriteriums (nach der WHO-Klassifikation von 2008) erbringen. Eine Knochenmark-Punktion ist nach den Diagnosekriterien der WHO von 2008 zur Diagnosestellung nur dann erforderlich, wenn nur das erste Major-Kriterium vorliegt und zwei Minor-Kriterien zum Beweis der Diagnose verlangt werden. Andererseits ist eine initiale Knochenmark-Punktion dennoch sinnvoll, da sie im Verlauf der Erkrankung eine Aussage über das Progressionsverhalten der Erkrankung und den Ausschluss einer frühen Fibrosierung des Knochenmarks erlaubt.
Für 2016 wird die Veröffentlichung der neuen WHO-Definition der PV erwartet. Vorschläge zu diesen neuen Diagnosekriterien sind aktuell schon bekannt [11]. So wurden die Hämoglobinwerte gesenkt, um zukünftig sogenannte „maskierte“ PV-Fälle frühzeitig diagnostizieren zu können (s. o.). Die Knochenmark-Punktion rückt zu den Major-Kriterien auf, erhält somit einen höheren Stellenwert und sollte nun obligatorisch durchgeführt werden. Eine Diagnosestellung in eindeutigen Fällen ist aber nach wie vor auch ohne Knochenmark-Punktion möglich. Das Kriterium der endogen erythroiden Kolonien wurde abgeschafft, da diese Untersuchung kaum standardisiert erhältlich war. Neue Untersuchungen wurden jedoch nicht in die neue WHO-Definition integriert, die notwendigen Untersuchungen bleiben somit die gleichen. Eine Gegenüberstellung der „alten“ und „neuen“ Diagnosekriterien findet sich in den Tabellen 1a und 1b.


Eine Sonografie des Abdomens ist neben der klinischen Einschätzung sinnvoll zur Bestimmung der initialen Milzgröße. Eine progrediente Splenomegalie kann unter anderem auf den beginnenden oder stattgehabten Übergang in eine Post-PV-Myelofibrose hinweisen und kann eine Indikation für eine zytoreduktive Therapie darstellen.
Differenzialdiagnosen
Unter den Differenzialdiagnosen der PV müssen die sogenannten primären und die wesentlich häufigeren sekundären Erythrozytosen (früher auch Polyglobulien genannt) unterschieden werden.
Zu den sekundären Erythrozytosen zählen in erster Linie die reaktiven Polyglobulien, welche bei nicht-eindeutigen Befunden mittels weiterer Tests (z. B. Lungenfunktions-Testung bei V. a. pulmonal verursachte Polyglobulie oder Echokardiografie bei V. a. kardiale Ursachen) ausgeschlossen werden sollten. Außerdem zeigen sich Polyglobulien bei Infektionen oder chronischen Entzündungen, die ebenfalls ausgeschlossen werden sollten. Eine Exsikkose oder Stress können ebenfalls ein PV-ähnliches Bild hervorrufen. Auch EPO-produzierende Nierentumoren oder Bronchialkarzinome können zu einer Erythrozytose führen, hierbei kann allerdings in Abgrenzung zur PV ein erhöhter Erythropoetin-Spiegel nachgewiesen werden.
Eine gestörte Erythropoetin-Sekretion oder auch eine Mutation im EPO-Rezeptor mit erniedrigter Affinität für EPO stellen weitere wichtige Differenzialdiagnosen dar. Außerdem können durch angeborene Mutationen im Hämoglobin-Protein (sogenannte Hämoglobinopathien) auch Erythrozytosen auftreten. Abbildung 1 fasst die Differenziadiagnostik der PV zusammen.
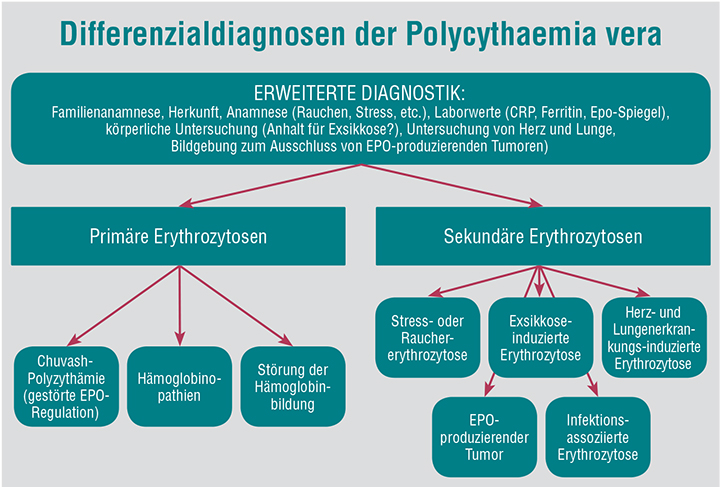
Weitere wichtige Differenzialdiagnosen sind die anderen MPN-Subentitäten. Bei der PV können alle drei Zellreihen zahlenmäßig erhöht sein, dies kann sich aber auch initial nur im Knochenmark und noch nicht im peripheren Blut widerspiegeln. Abgrenzungen zu den anderen MPN-Subentitäten lassen sich u. a. durch die zytomorphologische Begutachtung des Blutausstriches treffen, da hier neben der quantitativen Auswertung auch eine Aussage zum Leukozyten-Subtyp getroffen werden kann. Bei einer CML stehen zwar eher die Leukozyten mit einer Linksverschiebung im Vordergrund, aber auch bei der CML kann eine Erythrozytose auftreten. Eine ET zeichnet sich eher durch eine Vermehrung der Thrombozyten aus, seltener auch der Leukozyten. Die hyperplastische Phase einer primären Myelofibrose kann ebenfalls mit einer Vermehrung v. a. der Leukozyten und der Thrombozyten einhergehen. Bei nicht eindeutigen Fällen kann eine molekulare Analyse (z. B. durch den Nachweis von BCR-ABL bei der CML) oder eine Knochenmark-Punktion zur Eingrenzung bzw. Abgrenzung der Diagnose hinsichtlich der anderen MPN-Subentitäten hilfreich sein.
Die Post-PV-Myelofibrose stellt eine wichtige Komplikation bzw. späte Verlaufsform der PV dar. Sie zeichnet sich durch erniedrigte Hämoglobin-/Hämatokrit-Werte aus, meist ist keine Aderlass-Therapie oder zytoreduktive Therapie zur Kontrolle des Hämatokrits mehr notwendig. Häufig besteht eher eine Anämie. Meist geht diese Diagnose mit einer Splenomegalie sowie einer LDH-Erhöhung einher. Die Leukozyten- und Thrombozyten-Werte können normal, erniedrigt oder erhöht sein. Entscheidend für die Diagnosestellung bzw. die Abgrenzung zur PV ist der Grad der Fibrose des Knochenmarks. Eine Zusammenfassung der Diagnosekriterien für eine Post-PV-Myelofibrose findet sich in Tabelle 2.

Therapie der Polycythaemia vera, Prognose und Risikoeinteilung
Indikation zur Therapie
Alle Patienten mit PV sollten Aderlässe zur Einstellung des Hämatokrit-Wertes sowie eine Thrombembolie-Prophylaxe mittels ASS erhalten, wenn keine Kontraindikationen vorliegen. Eine solche Kontraindikation ist das sekundäre (erworbene) von Willebrand-Syndrom (sVWS). Dieses tritt insbesondere ab Thrombozyten-Werten von über 1.000 G/l auf, kann aber auch bei normwertigen Thrombozyten-Zahlen vorkommen. Die Gabe von ASS bei Patienten mit sVWS ist mit einem erhöhten Risiko für signifikante Blutungsereignisse vergesellschaftet [12].
Nach Sicherung der Diagnose einer PV sollte zunächst eine Risiko-Eingruppierung des individuellen Patienten erfolgen. Patienten im Alter von über 60 Jahren und solche, die bereits ein thromboembolisches Ereignis erlitten haben, haben ein deutlich erhöhtes Risiko für thromboembolische und schwergradige Blutungsereignisse. Daher stellen diese beiden Risikofaktoren eine Indikation für eine zytoreduktive Therapie dar.
In den neuen Leitlinien der DGHO von 2014 werden noch weitere Kriterien definiert, die eine Indikation für eine zytoreduktive Therapie darstellen können: Hierzu gehören ein koexistierender substitutionspflichtiger Eisenmangel, der Nachweis kardiovaskulärer Komorbiditäten oder eine progrediente PV (progrediente Splenomegalie, Thrombozyten-Werte > 600 G/l, Leukozyten-Werte > 25 G/l, hohe Aderlass-Frequenz oder leukoerythroblastisches Blutbild; [13]).
Allgemeine Therapieziele
Patienten mit PV, die einen anhaltend erhöhten Hämatokrit aufweisen, erleiden signifikant häufiger thromboembolische Komplikationen als solche, bei denen der Hämatokrit-Wert unter 45% gehalten wurde [14]. Daher sollte der Hämatokrit unter 45% gehalten werden, durch regelmäßige Aderlässe mit oder ohne zytoreduktive Therapie. Bei Frauen wird aufgrund der geringeren Körpermasse und des dadurch verringerten Blutvolumens von einigen Autoren ein Zielwert unter 42% diskutiert [15], der sich jedoch bisher nicht durchgesetzt hat. Die Frequenz der notwendigen Aderlässe lässt mit der Zeit durch die zunehmende Entleerung der Eisenspeicher meistens nach. Ein Eisenmangel sollte bei der PV nur in Ausnahmefällen (z. B. bei schwergradiger extra-hämatopoetischer Eisenmangel-Symptomatik) ausgeglichen werden, da eine Eisengabe die Erythrozyten-Produktion anregt und der Therapie der PV so entgegenwirkt. Durch die Erniedrigung des Hämatokrit-Wertes unter 45% kann das Risiko für thromboembolische Ereignisse gegenüber einer Einstellung auf Werte zwischen 45% und 50% um das Vierfache reduziert werden.
Therapeutika
Hydroxyurea
Bis heute ist Hydroxyurea das einzige zugelassene Medikament für die Erstlinien-Therapie der PV. In einer retrospektiven Analyse von Alvarez-Larran zeigte sich bei 261 Patienten eine gute Verträglichkeit und eine komplette Remissionsrate von 24% [16]. Unter Hydroxyurea konnte eine gute Hämatokrit- und Symptomkontrolle sowie eine gute Wirkung auf die Splenomegalie gezeigt werden. Allerdings wird Hydroxyurea aufgrund des nicht auszuschließenden leukämogenen Potenzials sowie des bekannten teratogenen Potenzials ungern langfristig bei jungen Patienten eingesetzt.
Ruxolitinib
Aufgrund der Daten der sogenannten RESPONSE-Studie, die die Wirksamkeit und Verträglichkeit von Ruxolitinib im Vergleich zur Standardtherapie bei Patienten mit Hydroxyurea-intoleranter oder -resistenter PV untersuchte [17], wurde Ruxolitinib im Sommer 2015 in Deutschland für die Behandlung der HU-intoleranten oder -refraktären PV zugelassen. In der Studie führte die Therapie mit Ruxolitinib zu einer stärkeren Reduktion der Milzgröße und einer besseren Hämatokrit-Kontrolle gegenüber der besten verfügbaren Therapie (21% vs. 1%; p < 0,001). Das Nebenwirkungsprofil umfasst vor allem hämatologische Nebenwirkungen wie Thrombozytopenie und Anämie, zudem ist vermehrt auf Infektionen zu achten. Insbesondere das Wiederaufflammen chronischer Infektionskrankheiten wie Hepatitis oder Tuberkulose wurde beschrieben, weshalb die Patienten vor Behandlung darauf untersucht und ggf. engmaschig monitoriert werden sollten.
Interferon
Die Interferon-Behandlung ist eine sehr effektive Therapie mit dem Potenzial, bei Patienten mit PV molekulare Remissionen zu induzieren. Die Therapie war aber in der Vergangenheit vor allem aufgrund der damals verwendeten hohen Dosen mit einer hohen Toxizität vergesellschaftet. Seit Einführung des pegylierten Interferons, welches aufgrund einer längeren Halbwertszeit nur noch einmal pro Woche appliziert werden muss, ist die Verträglichkeit der Substanz deutlich gestiegen, während die Ansprechraten zwischen 29% und 97% liegen [18]. Aktuell sind jedoch weder das konventionelle noch das pegylierte Interferon in Deutschland für die Behandlung der PV zugelassen. Nichtsdestotrotz wird die Interferon-Therapie aufgrund der sehr guten Ansprechraten und der Möglichkeit der molekularen Remission in den aktuellen Leitlinien der DGHO empfohlen. Aktuell wird im Rahmen von klinischen Studien ein weiteres Interferon-Derivat (sogenanntes Ropeginterferon alfa-2b) getestet, welches nur alle zwei Wochen injiziert werden muss. Erste Daten sind vielversprechend, in einer Studie mit 51% PV-Patienten konnte eine 90%ige Gesamtansprechrate erreicht werden [19].
Experimentelle Ansätze
Derzeit wird eine orale Verabreichungsform von pegyliertem Interferon bei Patienten mit PV im Rahmen einer Phase-I-Studie [NCT02407080] getestet. Des Weiteren läuft eine Phase II-Studie, welche das Beta3-Sympathomimetikum Mirabegron bei JAK2-positiven MPN testet (NCT02311569), nachdem diese Substanzklasse in präklinischen Untersuchungen positive Auswirkungen auf die MPN-Stammzellnische gezeigt haben. Auch Inhibitoren von Histondeacetylasen (HDAC-Inhibitoren) werden aktuell im Rahmen von Studien bei der PV überprüft. In einer multizentrischen Phase-II-Studie wurden 44 Patienten mit HU-intoleranter oder HU-refraktärer PV mit dem HDAC-Inhibitor Givinostat in Kombination mit HU behandelt. Dabei zeigten sich eine Gesamtansprechrate von 50% bzw. 55% (je nach Therapiearm unter 50 oder 100 mg Givinostat). Die Kombination war gut verträglich [20].
Diese Substanzen müssen noch weiter evaluiert werden, um genaue Aussagen über die Wirksamkeit und Verträglichkeit zu erlauben. Auch Kombinationstherapien werden aktuell im Rahmen von Studien getestet. So wurde bereits in einer kleinen Kohorte die Wirksamkeit und Verträglichkeit einer Kombinationstherapie aus Interferon und JAK-Inhibitor getestet. Hier führte die Kombination aus Interferon alfa-2a und Ruxolitinib innerhalb von vier Wochen zu einer Normalisierung der Blutwerte, und in zehn Monaten reduzierte sich die JAK2-Allellast von 90% auf 28% [21].
Therapieansprechen
Die Empfehlungen zur Evaluation des Ansprechens auf eine zytoreduktive Therapie bei PV haben sich zuletzt 2013 [22, 23] signifikant geändert. Zum Nachweis einer kompletten Remission ist nunmehr eine Knochenmark-Punktion notwendig. Eine Gegenüberstellung der alten und neuen Kriterien ist in Tabelle 3 dargestellt.

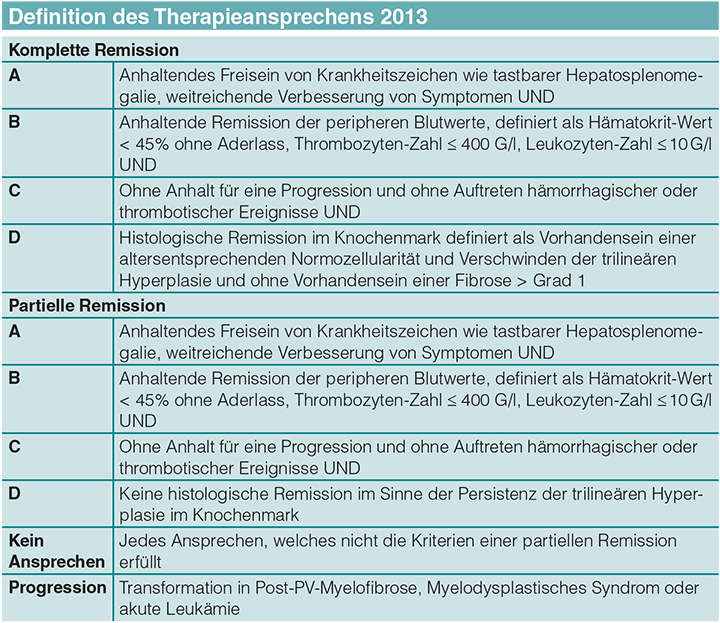
Der Stellenwert dieser Kriterien ist derzeit umstritten, da eine Therapie der PV eher auf eine Symptomkontrolle und das Vermeiden thromboembolischer Ereignisse abzielt [16]. Ein Vorteil für Patienten bei Erreichen einer kompletten Remission im Sinne von Verlängerung des Gesamtüberlebens oder ähnlichem konnte bislang nicht nachgewiesen werden. Andererseits wird die klare Festlegung und Anwendung allgemeiner Remissionskriterien international gefordert, da in den letzten Jahren Studienendpunkte oft sehr substanzspezifisch gewählt wurden (z. B. 35%ige Reduktion des Milzvolumens etc.) und somit der Vergleich mehrerer Substanzen, wenn sie nicht randomisiert im Rahmen derselben Studie getestet wurden, praktisch nicht möglich ist.
Literatur
1. Barbui T et al. Masked polycythemia vera diagnosed according to WHO and BCSH classification. Am J Hematol 2014; 89: 199-202.
2. Johansson P. Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. Semin Thromb Hemost, 2006; 32: 171-3.
3. Leonard WJ. Role of Jak kinases and STATs in cytokine signal transduction. Int J Hematol 2001; 73: 271-7.
4. Stein BL et al. Polycythemia vera: An appraisal of the biology and management 10 years after the discovery of JAK2 V617F. J Clin Oncol 2015, Aug 31 [Epub ahead of print, pii: JCO.2015.61.6474].
5. Spivak JL et al. Two clinical phenotypes in polycythemia vera. N Engl J Med 2014; 371: 808-17.
6. Ortmann CA et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med 2015; 372: 601-12.
7. Scherber R et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): International prospective validation and reliability trial in 402 patients. Blood 2011; 118: 401-8.
8. Siegel FP et al. Aquagenic pruritus in polycythemia vera: Characteristics and influence on quality of life in 441 patients. Am J Hematol 2013; 88: 665-9.
9. Polycythemia vera: the natural history of 1213 patients followed for 20 years. Gruppo Italiano Studio Policitemia. Ann Intern Med 1995; 123: 656-64.
10. Swerdllow SH et al. WHO classification of tumours of haematopoietic and lymphoid tissues. France: IARC Press, 2008.
11. Tefferi A et al. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia 2014; 28: 1407-13.
12. Griesshammer M et al. Aspirin in essential thrombocythemia: Status quo and quo vadis. Semin Thromb Hemost 1997; 23: 371-7.
13. Lengfelder E, Baerlocher GM, Gisslinger H, Grießhammer M, Petrides PE. Polycythaemia Vera (PV). Onkopedia Leitlinien 2014 [https://www.onkopedia.com/de/onkopedia/guidelines/polycythaemia-vera-pv/@@view/html/index.html].
14. Marchioli R et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013; 368: 22-33.
15. Spivak JL. Polycythemia vera, the hematocrit, and blood-volume physiology. N Engl J Med 2013; 368: 76-8.
16. Alvarez-Larran A et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012; 119: 1363-9.
17. Vannucchi AM. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med 2015; 372: 1670-1.
18. Kiladjian JJ et al. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011; 117: 4706-15.
19. Gisslinger H et al. Ropeginterferon alfa-2b, a novel IFNalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood 2015; 126: 1762-9.
20. Finazzi G et al. A phase II study of givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br J Haematol 2013; 161: 688-94.
21. Bjorn ME et al. Combination therapy with interferon and JAK1-2 inhibitor is feasible: Proof of concept with rapid reduction in JAK2V617F-allele burden in polycythemia vera. Leuk Res Rep 2014; 3: 73-5.
22. Barosi G et al. Response criteria for essential thrombocythemia and polycythemia vera: Result of a European LeukemiaNet consensus conference. Blood 2009; 113: 4829-33.
23. Barosi G et al. Revised response criteria for polycythemia vera and essential thrombocythemia: An ELN and IWG-MRT consensus project. Blood 2013; 121: 4778-81.

Korrespondierende Autorin:
Dr. med. Susanne Isfort
Klinik für Hämatologie, Onkologie, Hämostaseologie und Stammzelltransplantation
Medizinische Fakultät der RWTH Aachen
Pauwelsstraße 30, 52074 Aachen
+49 241 8037411
+49 241 8082449
sisfort[at]ukaachen[dot]de

Prof. Dr. med. Tim H. Brümmendorf
Prof. Dr. med. Steffen Koschmieder
Klinik für Hämatologie, Onkologie, Hämostaseologie und Stammzelltransplantation
Medizinische Fakultät der RWTH Aachen