Essenzielle Thrombozythämie (ET)
Frank Stegelmann, Konstanze Döhner
Die essenzielle Thrombozythämie (ET) ist eine chronische myeloische Stammzellerkrankung, bei der vor allem die Megakaryozyten von einer autonomen Proliferation betroffen sind. Folge ist eine Thrombozytose im peripheren Blut. Neben der Thrombopoese kann zusätzlich auch die Granulopoese gesteigert sein. Die Inzidenz der ET ist gering und liegt bei etwa 1–2,5 pro 100.000 Einwohner pro Jahr [1]. Durch die insgesamt günstige Prognose und den langen Krankheitsverlauf liegt ihre Prävalenz jedoch weit höher als die Inzidenz vermuten lässt. Die Erkrankung wird meistens zwischen dem 50. und 60. Lebensjahr festgestellt. Aufgrund häufiger Blutbildbestimmungen wie z. B. im Rahmen von Vorsorgeuntersuchungen wird die ET aber zunehmend auch bei jüngeren Patienten diagnostiziert: Etwa 20% aller Patienten sind jünger als 40 Jahre. Gelegentlich sind auch Kinder oder junge Erwachsene betroffen.
Differenzialdiagnose und Diagnosefindung
Differenzialdiagnostisch steht die Abgrenzung der ET-bedingten Thrombozytose von den häufigeren reaktiven Ursachen im Vordergrund. Beispiele hierfür sind ein Eisenmangel bei okkultem oder nach akutem Blutverlust oder auch entzündliche Prozesse im Rahmen von Infektions-, Tumor- oder Autoimmunerkrankungen. Darüber hinaus ist die Abgrenzung der ET von anderen hämatologischen Erkrankungen von Bedeutung. In diesem Zusammenhang ist insbesondere die Myelofibrose (MF) im präfibrotischen Stadium zu nennen. Diese geht häufiger in eine manifeste Knochenmarks-Fibrose über als die ET und ist daher langfristig mit einer ungünstigeren Prognose assoziiert [2]. Die präfibrotische MF ist wie die ET durch eine meist isolierte Thrombozytose des peripheren Bluts gekennzeichnet, die oft höher ausgeprägt ist als bei der „wahren“ ET. Hämoglobin-Konzentration und Leukozyten sind meist normwertig. Zeichen der manifesten MF wie z. B. eine ausgeprägte Splenomegalie und ein leukoerythroblastisches Blutbild fehlen in der Regel. Lediglich der LDH-Wert im Blut ist bei der präfibrotischen MF häufiger erhöht als bei der ET. Eine sichere Abgrenzung kann jedoch nur anhand der Knochenmarkhistologie durch einen erfahrenen Pathologen erfolgen. Während bei der ET die Megakaryozyten im Knochenmark typischerweise vergrößert, hyperlobuliert und locker in Gruppen gelagert sind, sind für die präfibrotische MF größenvariable, oftmals kleine und dicht gelagerte Megakaryozyten mit irregulärer Kernstruktur charakteristisch [3].
In den letzten Jahren gelang bei myeloproliferativen Neoplasien die Identifizierung von spezifischen Genmutationen. Dies erleichtert die Diagnosestellung der ET erheblich. In etwa 50–60% der Fälle findet sich die aktivierende Punktmutation V617F im Gen für die Janus-Kinase-2 (JAK2; [4–7]), bei weiteren 20–30% der Fälle eine Frameshift-Mutation im Calreticulin-Gen (CALR; [8–9]), und ca. 5% der Patienten tragen eine Punktmutation im Gen für den Thrombopoetinrezeptor MPL (W515L/K; [10]). Da die drei genannten Mutationen überwiegend unabhängig voneinander vorkommen, kann durch eine PCR-basierte Testung der drei Marker bei 80–90% der ET-Patienten eine Mutation nachgewiesen werden. Standard hierfür ist Leukozyten- bzw. Granulozyten-DNA aus dem peripheren Blut, anhand derer die Klonalität der Erkrankung nachweisbar ist. Eine Knochenmark-Untersuchung ist hierfür nicht notwendig. Gemäß den aktuellen WHO-Empfehlungen [3] sollte die Diagnose der ET in Zusammenschau von Laborwerten, Knochenmark-histologischem Bild und – wenn nachweisbar – einer Genmutation (JAK2/CALR/MPL) gestellt werden (Tab. 1).

Klinisches Erscheinungsbild und Komplikationen
Mit der Dauer der Erkrankung steigt das Risiko für eine Veränderung des klinischen Erscheinungsbildes der ET. Ein Übergang in eine Polycythaemia vera (PV) oder in eine sekundäre akute myeloische Leukämie ist jedoch selten und kommt retrospektiven Studien zufolge bei deutlich weniger als 5% der Patienten nach 15 Jahren Beobachtungszeit vor [2]. Etwas häufiger ist eine Transformation in die sogenannte Post-ET MF. Die Häufigkeit fibrotischer Transformationen beträgt bei der ET ca. 5–10% nach 15 Jahren. Das Gesamtüberleben der ET-Patienten liegt nach dieser Zeit bei etwa 80% und ist damit gegenüber der gesunden Normalbevölkerung nur leicht eingeschränkt. Problematisch ist die ET daher – langfristig gesehen – vor allem für jüngere Patienten mit noch sehr langer Lebenserwartung. Um ein optimales Management sicherzustellen, sollten ET-Patienten daher in jedem Fall von hämatologischen Fachkollegen (mit-)betreut werden.
Das klinische Erscheinungsbild der ET ist insgesamt sehr variabel. Etwa ein Drittel der Fälle ist zum Zeitpunkt der Diagnosestellung asymptomatisch. Die Diagnose wird in diesen Fällen als Folge einer zufällig festgestellten Thrombozytose im Rahmen einer Blutbilduntersuchung gestellt. Ein weiteres Drittel der Patienten klagt bei Erstdiagnose über Missempfindungen der Akren (Finger und/oder Zehen), über auf Analgetika häufig kaum ansprechende Kopfschmerzen oder über selbstlimitierende Episoden von Schwindel, Seh- oder Hörstörungen ohne anderweitige Ursache. Diese Symptome werden auch unter dem Begriff Mikrozirkulationsstörungen zusammengefasst. Ihre Ursache liegt in einer durch die Thrombozytose bedingten gestörten Kapillar-Durchblutung.
Bei etwa einem weiteren Drittel der Patienten wird die Diagnose der ET in der Folge eines manifesten Gefäßverschlusses gestellt. Arterien sind dabei häufiger betroffen als das venöse System [11]. Typisch sind thromboembolische Verschlüsse von zerebralen, kardialen oder peripher-arteriellen Gefäßen. Typische Komplikationen im venösen System sind oberflächliche und tiefe Venenthrombosen. Neben den Beinvenen sind vor allem viszerale Thrombosen (z. B. Pfortader- oder Milzvenenthrombose, Budd-Chiari-Syndrom) oder auch eine Sinusvenenthrombose nicht seltene Manifestationsorte. Blutungsereignisse hingegen entstehen vor allem in Verbindung mit sehr hohen Thrombozytenwerten (meist > 1.000–1.500 x 109/l) und einem konsekutiv erworbenen von Willebrand-Syndrom [12]. Im Verlauf liegt die Thrombosehäufigkeit der ET im Mittel bei etwa 10–20% nach zehn Jahren und kann durch ein entsprechendes Management gesenkt werden.
Risiko-Nutzen-Abwägung der Zytoreduktion
Die Abwägung des Nutzens einer medikamentösen Behandlung gegenüber den – vor allem in der Langzeittherapie – zu erwartenden bzw. möglichen Nebenwirkungen ist bei der ET essenziell. Grundlage dafür ist eine sorgfältige und individuelle Abschätzung des Risikos für das Auftreten von Gefäßkomplikationen, die die Therapie rechtfertigt. Die Datenlage aus prospektiven, kontrollierten Studien ist bei der ET überschaubar. Darüber hinaus wurde das Management aber auch durch eine Reihe von retrospektiven Analysen gut charakterisierter Patientenkollektive in den letzten Jahren beeinflusst.
Als wichtigste Risikofaktoren für die Initiierung einer medikamentösen Therapie gelten bei der ET ein Alter von über 60 Jahren und ein stattgehabtes thromboembolisches Ereignis, das mit der ET in Zusammenhang steht [13]. Das Vorliegen von einem der beiden Faktoren ist bereits ausreichend für die Einstufung als Hochrisiko-ET. In diesem Fall ist eine zytoreduktive Behandlung indiziert mit dem Ziel, eine dauerhafte Normalisierung des peripheren Thrombozyten-Werts zur Senkung des vaskulären Risikos zu erreichen (Abb. 1).
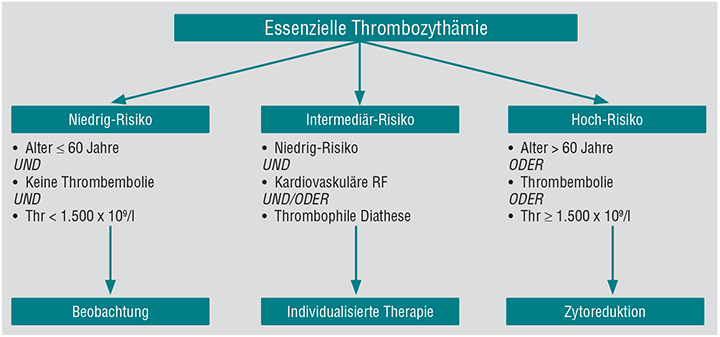
Des Weiteren gelten Patienten mit Thrombozyten-Werten > 1.500 x 109/l aufgrund des signifikant erhöhten Blutungsrisikos als behandlungsbedürftig. Obwohl keine eindeutigen Studiendaten in dieser Situation vorliegen, ist bei diesen Patienten ebenfalls eine Senkung des Thrombozyten-Werts in den Normbereich anzustreben. Patienten ≤ 60 Jahre ohne vorangegangenes Gefäßereignis und mit Thrombozyten-Werten < 1.500 x 109/l werden dagegen als Niedrigrisiko-ET eingestuft und müssen nicht behandelt werden. Es wird empfohlen, auf das Vorliegen und ein entsprechendes Management begleitender kardiovaskulärer Risikofaktoren (z. B. Rauchen, arterielle Hypertonie, Hypercholesterinämie, Diabetes mellitus, Adipositas) zu achten.
Diese Patienten werden häufig in eine weitere Risikogruppe, die sogenannte Intermediärrisiko-ET, eingestuft. Dies gilt auch für andere thrombophile Risikofaktoren wie z. B. eine Faktor V-Leiden-Mutation, eine Prothrombinogen-Mutation, einen Protein-C- oder Protein-S-Mangel, einen Antithrombin-III-Mangel oder ein Antiphospholipid-Syndrom. In diesen Fällen muss eine individuelle Entscheidung getroffen werden, ob eine zytoreduktive Behandlung zur Normalisierung der Thrombozyten sinnvoll ist. Allgemeingültige Empfehlungen existieren hierzu nicht.
Neben den drei im Management der ET fest etablierten Risikofaktoren (Alter über 60 Jahre, stattgehabtes Gefäßereignis, Thrombozyten > 1.500 x 109/l) gewinnen auch Untersuchungen zu den jeweiligen molekulargenetischen Subgruppen eine zunehmende Bedeutung. Während eine JAK2 V617F-Mutation mit einem tendenziell höheren Risiko für das Auftreten von vaskulären Ereignissen assoziiert ist, werden CALR- und MPL-Mutationen bei den Patienten nicht mit
einem ungünstigeren Verlauf in Verbindung gebracht [11, 14]. Die Datenlage reicht allerdings bislang nicht aus, vom Vorliegen einer JAK2-Mutation eine Therapieentscheidung abhängig zu machen.
Der Nutzen neuerer Risiko-Scores wie z. B. des „IPSET-Thrombosis“ (Internationaler Prognose-Score bei WHO-definierter ET), in den kardiovaskuläre Risikofaktoren und die JAK2 V617F-Mutation miteingehen, ist durch fehlende prospektive Validierung eingeschränkt [15]. Daher ist z. B. die daraus hervorgegangene Empfehlung, ET-Patienten im Alter von über 60 Jahren, die keine JAK2-Mutation und keine weiteren Risikofaktoren aufweisen, nicht zu behandeln, kritisch zu hinterfragen.
Ähnliche Unklarheit besteht auch für die Leukozytenzahl der ET-Patienten. Eine Leukozytose wurde in retrospektiven Untersuchungen ebenfalls mit einem erhöhten vaskulären Risiko in Verbindung gebracht [11]. Dies sollte aber die Initiierung einer zytoreduktiven Therapie nicht alleine rechtfertigen, sondern im Kontext mit anderen Risikofaktoren kritisch beleuchtet werden.
Therapie der ET
Hydroxyurea und Anagrelid: zugelassene Therapieoptionen
Zur zytoreduktiven Behandlung sind bei der ET zwei Medikamente zugelassen, das orale Zytostatikum Hydroxyurea (HU) und Anagrelid, ein selektiver Thrombozyten-Reduktor. HU steht hierbei ab der ersten Therapielinie, Anagrelid gemäß Zulassungstext ab der Zweitlinientherapie zur Verfügung. In der 2005 publizierten PT1-Studie mit 809 Hochrisiko-ET-Patienten war Anagrelid im randomisierten Vergleich mit HU hinsichtlich einer Reduktion von arteriellen Gefäßereignissen, nicht aber hinsichtlich einer Reduktion von venösen Thrombosen signifikant unterlegen [16]. Zudem traten im Anagrelid-Arm mehr Blutungsereignisse auf. Problematisch bei der Interpretation dieser Ergebnisse ist die Tatsache, dass alle Patienten begleitend mit niedrigdosierter Azetylsalizylsäure (ASS) behandelt wurden. In der gleichermaßen randomisierten ANAHYDRET-Studie mit 259 ET-Patienten ohne begleitende ASS-Therapie konnten diese Ergebnisse nicht bestätigt werden [17]. Die Studie war auf Nicht-Unterlegenheit von Anagrelid ausgelegt und zeigte keinen signifikanten Unterschied der beiden Therapiearme hinsichtlich des Auftretens von thromboembolischen oder hämorrhagischen Komplikationen. Somit stehen mit HU und Anagrelid zwei nahezu gleichwertige Medikamente mit guter Wirksamkeit zur Vermeidung vaskulärer Komplikationen bei der Hochrisiko-ET zur Verfügung.
HU sollte aufgrund seiner unspezifisch-zytostatischen Eigenschaften bei jüngeren Patienten (v. a. unter 40 Jahren) zurückhaltend eingesetzt werden [13]. Typische Nebenwirkungen von HU sind hämatologische (Anämie/Leukopenie) und mukokutane Toxizität. Letztere erstreckt sich von gastrointestinalen Beschwerden und Hautrötungen bzw. Lichtempfindlichkeit der Haut über mukokutane Ulzera bis hin zu aktinischen Keratosen oder dem Auftreten von Basaliomen und Plattenepithelkarzinomen. Diese treten vor allem an der lichtexponierten Haut auf. Das Auftreten solcher Nebenwirkungen ist dosis- und zeitabhängig und nicht mit exakten Zahlen belegt.
Die Abbruchrate von Anagrelid aufgrund von Nebenwirkungen liegt nach zwei Jahren bei bis zu 50% [18]. Häufigste Nebenwirkungen dieses Medikaments sind Kopfschmerzen, supraventrikuläre Tachykardien und Magen-Darm-Beschwerden. Nebenwirkungen können unter Umständen durch eine möglichst gleichmäßig über den Tag verteilte Einnahme des Medikaments gelindert werden.
In vielen Fällen kann auch eine Kombination von HU und Anagrelid sinnvoll sein, um die Dosen beider Medikamente zu reduzieren und somit ein besseres Nebenwirkungsmanagement zu ermöglichen. HU wird in der Regel mit einer ein- bis zweimal täglichen Einnahme von 500 mg begonnen, während die Anfangsdosis von Anagrelid bei 0,5 mg ein- bis zweimal täglich liegt. In Abhängigkeit vom Thrombozyten-Verlauf sind entsprechende Dosisanpassungen beider Medikamente individuell vorzunehmen. Die tägliche HU-Dosis sollte möglichst 2.000 mg nicht überschreiten, die maximale Tagesdosis von Anagrelid liegt bei 5 mg.
Datenlage zu Interferon-α
Interferon-α (IFN-α) ist ebenfalls ein gut wirksames Medikament bei der ET und wird seit vielen Jahren zur Zytoreduktion bei Hochrisiko-Patienten eingesetzt. Für diese Substanz gibt es jedoch trotz der langjährigen Erfahrungen nach wie vor keine randomisierten Daten im Vergleich mit einer etablierten Standardtherapie wie HU oder Anagrelid, sodass das Medikament bei der ET nicht zugelassen ist. Nach gegenwärtigen Empfehlungen ist eine IFN-α-Therapie – nach einer entsprechenden Zusage der Kostenübernahme durch den Versicherungsträger – vor allem bei jüngeren Patienten mit Versagen oder Intoleranz gegenüber Anagrelid indiziert [13]. Die pegylierte Form von IFN-α (Peg-IFN-α2a oder -α2b) ist dabei hinsichtlich der Häufigkeit von Nebenwirkungen konventionellem IFN-α überlegen und ermöglicht aufgrund seiner Pharmakokinetik eine nur einmal wöchentliche subkutane Applikation.
Interessanterweise birgt IFN-α das Potenzial, nicht nur die Thrombozyten im Sinne einer hämatologischen Remission zu normalisieren, sondern auch zu einer Reduktion des malignen Zellklons zu führen und damit, ähnlich wie bei der chronischen myeloischen Leukämie, eine Remission auf zytomorphologischer bzw. molekularer Ebene zu erreichen. Dies wurde anhand von seriell durchgeführten Knochenmark-Histologien sowie bei JAK2 V617F-mutierten ET-Patienten mittels serieller quantitativer PCR-Messungen gezeigt [19–20]. Etwa 20–30% der Patienten erreichen nach mehrjähriger Behandlung mit IFN-α eine molekulare Remission mit Rückgang der JAK2-Allellast auf 1% oder weniger. Möglicherweise kann somit neben einer Verminderung des Risikos für vaskuläre Ereignisse auch eine langfristige Krankheitsmodifikation erreicht werden, die die Progressionsrate (fibrotische und/oder leukämische Transformationen) signifikant senkt. Dies ist Gegenstand aktueller wissenschaftlicher Untersuchungen und vor allem für jüngere ET-Patienten von enormer Relevanz.
Im Gegensatz zu HU und Anagrelid ist die mediane Zeit bis zum hämatologischen Ansprechen bei IFN-α länger. In einer Phase-II-Studie mit Peg-IFN-α2b war z. B. eine Normalisierung der Thrombozytenzahl bei etwa 37% der Patienten nach drei Monaten erreicht, und erst nach zwölf Monaten Therapie lag bei 67% eine hämatologische Remission vor [21]. Die Startdosis des Medikaments betrug dabei 50 µg pro Woche und entsprach auch der medianen Dosierung im weiteren Verlauf.
Als häufigste Nebenwirkungen treten unter IFN-α grippeartige Symptome (Fieber, Schüttelfrost, Kopf- und Gliederschmerzen) nach der Applikation ein, die einige Stunden bis wenige Tage persistieren können und durch begleitende Paracetamol-Einnahme in vielen Fällen gut behandelbar sind. Des Weiteren kann die Therapie in selteneren Fällen eine Depression oder eine Autoimmunerkrankung induzieren bzw. diese verschlechtern, weshalb eine IFN-α-Therapie bei Patienten mit entsprechender Komorbidität kontraindiziert ist. Die Abbruchrate bei pegyliertem IFN-α liegt nach zwei Jahren bei etwa einem Drittel. In der dauerhaften Anwendung über Jahre hinweg ist die Therapie mit dem Medikament bei etwa einem Drittel der Patienten durchführbar, zwei Drittel brechen die Therapie – meist aufgrund von Nebenwirkungen – im Laufe der Zeit ab.
Interessanterweise wurden in den letzten Jahren zunehmend Fälle beschrieben, die auch nach Absetzen des Medikaments keine erneute Zytoreduktion benötigen und in hämatologischer Remission bleiben [22]. Ursache dafür ist möglicherweise eine anhaltende Immunstimulation, deren exakte Mechanismen bislang allerdings unzureichend erforscht sind.
Stellenwert von Azetylsalizylsäure
Der Stellenwert einer Thrombozyten-Aggregationshemmung mit niedrigdosiertem ASS (50–100 mg pro Tag) ist bei der ET im Gegensatz zur PV, bei der dies neben der Hämatokrit-Senkung ein fester Bestandteil der Dauertherapie ist, unklar. Belegt ist lediglich, dass ASS als symptomatische Therapie bei ET-bedingten Mikrozirkulationsstörungen von Nutzen ist. Von wahrscheinlichem Nutzen hinsichtlich der Protektion vor vaskulären Komplikationen ist der Einsatz von ASS auch bei stattgehabten arteriellen Ereignissen. Bei anamnestisch bekannter hämorrhagischer Diathese oder Ulkusleiden ist ASS kontraindiziert. Außerdem sollte ASS bei sehr hohen Thrombozyten-Werten (> 1.000–1.500 x 109/l) nicht mehr verabreicht werden, da es in dieser Situation die Blutungsneigung im Rahmen eines erworbenen von Willebrand-Syndroms verstärken und somit zu Blutungen führen kann [12].
JAK- und Telomerase-Inhibitoren: Experimentelle Substanzen
Zum JAK1/2-Inhibitor Ruxolitinib, der bei der MF und in der Zweitlinientherapie der PV mittlerweile zugelassen ist und krankheitsassoziierte Symptome mindert bzw. eine Hämatokrit-Kontrolle ohne Aderlässe ermöglicht, existieren bei der ET nur wenige Studiendaten. Erste Ergebnisse einer Phase-I/II-Studie mit Ruxolitinib in verschiedenen Dosisstufen von 20 mg bis 50 mg täglich zeigen relativ geringe Ansprechraten bezüglich Reduktion bzw. Normalisierung der Thrombozyten. So wurde in dieser Studie bei 38 ET-Patienten nur in einem Fall eine partielle hämatologische Remission berichtet [23]. Eine endgültige Publikation der Daten steht jedoch aus.
In Kürze wird in Deutschland die sog. Ruxo-BEAT-Studie zur Verfügung stehen, in die neben PV- auch unbehandelte und vorbehandelte Hochrisiko-ET-Patienten eingeschlossen werden können. In dieser Studie wird eine Therapie mit Ruxolitinib gegenüber anderen verfügbaren zytoreduktiven Therapien randomisiert geprüft. Primärer Endpunkt ist die Rate kompletter hämatologischer Remissionen nach sechs Monaten Therapie. Leiter der klinischen Prüfung ist Herr Professor Dr. Koschmieder vom Universitätsklinikum Aachen, Sponsor ist die RWTH Universität Aachen. Ob JAK-Inhibitoren, die sich derzeit in der klinischen Entwicklung befinden, künftig Eingang in die Therapie der ET erhalten werden, ist derzeit noch nicht absehbar.
Darüber hinaus wurden kürzlich interessante Daten zum Einsatz des Telomeraseinhibitors Imetelstat publiziert [24]. Bei der Substanz handelt es sich um ein Lipid-modifiziertes Oligonukleotid, das die enzymatische Aktivität der Telomerase inhibiert und einmal wöchentlich intravenös verabreicht werden muss. In einer Phase-II-Studie bei 18 ET-Patienten lag die Rate kompletter hämatologischer Remissionen bei 89%. Toxizitäten, die beobachtet wurden, waren vor allem Leukopenie/Anämie, Leberwert-Erhöhungen und Kopfschmerzen. Die Substanz wurde jedoch als insgesamt gut verträglich beschrieben. Interessanterweise ist das schnelle hämatologische Ansprechen auch von einer deutlichen Reduktion der Allellast, gemessen mittels quantitativer Bestimmungen von mutiertem JAK2, MPL und CALR, bei den meisten Patienten begleitet. Inwieweit Imetelstat Einzug in die Therapie der ET halten wird und den Erkrankungsverlauf positiv beeinflussen kann, muss noch mittels einer Phase-III-Studie mit einer größeren Patientenzahl untersucht werden.
Literatur
1. Mesa RA et al. Population-based incidence and survival figures in essential thrombocythemia and agnogenic myeloid metaplasia: An Olmsted County Study, 1976-1995. Am J Hematol 1999; 61: 10-5.
2. Barbui T et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: An international study. J Clin Oncol 2011; 29: 3179-84.
3. Vardiman JW et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009; 114: 937-51.
4. Kralovics R et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779-90.
5. James C et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434: 1144-8.
6. Baxter EJ et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365: 1054-61.
7. Levine RL et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7: 387-97.
8. Pikman Y et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006; 3: e270.
9. Klampfl T et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013; 369: 2379-90.
10. Nangalia et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013; 369: 2391-405.
11. Carobbio A et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: An international study of 891 patients. Blood 2011; 117: 5857–9.
12. Michiels JJ et al. The paradox of platelet activation and impaired function: Platelet-von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in essential thrombocythemia and polycythemia vera. Semin Thromb Hemost 2006; 32: 589–604.
13. Barbui T et al. Philadelphia-negative classical myeloproliferative neoplasms: Critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011; 29: 761-70.
14. Finazzi G et al. Calreticulin mutation does not modify the IPSET score for predicting the risk of thrombosis among 1150 patients with essential thrombocythemia. Blood 2014; 124: 2611–2.
15. Finazzi G et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood 2012; 120: 5128-33.
16. Harrison CN et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005; 353: 33-45.
17. Gisslinger H et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: The ANAHYDRET Study, a randomized controlled trial. Blood 2013; 121: 1720-8.
18. Besses C et al. Cytoreductive treatment patterns for essential thrombocythemia in Europe. Analysis of 3643 patients in the EXELS study. Leuk Res 2013; 37: 162-8.
19. Quintás-Cardama A et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 2009; 27: 5418-24.
20. Stauffer Larsen T et al. Long term molecular responses in a cohort of Danish patients with essential thrombocythemia, polycythemia vera and myelofibrosis treated with recombinant interferon alpha. Leuk Res 2013; 37: 1041-5.
21. Langer C et al. Pegylated interferon for the treatment of high risk essential thrombocythemia: Results of a phase II study. Haematologica 2005; 90: 1333-8.
22. Utke Rank C et al. Minimal residual disease after long-term interferon-alpha2 treatment: A report on hematological, molecular and histomorphological response patterns in 10 patients with essential thrombocythemia and polycythemia vera. Leuk Lymphoma 2015; [Epub ahead of print].
23. Pieri L et al. JAK2V617F complete molecular remission in polycythemia vera/essential thrombocythemia patients treated with ruxolitinib. Blood 2015; 125: 3352-3.
24. Baerlocher GM et al. Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N Engl J Med 2015; 373: 920-8.

(korrespondierender Autor)
Dr. med. Frank Stegelmann
Prof. Dr. med. Konstanze Döhner
Klinik für Innere Medizin III, Zentrum für Innere Medizin; Universitätsklinikum Ulm
Albert-Einstein-Allee 23, 89081 Ulm
frank.stegelmann[at]uniklinik-ulm[dot]de