Periphere T-/NK-Zell-Lymphome
Peter Reimer
Epidemiologie und Einteilung
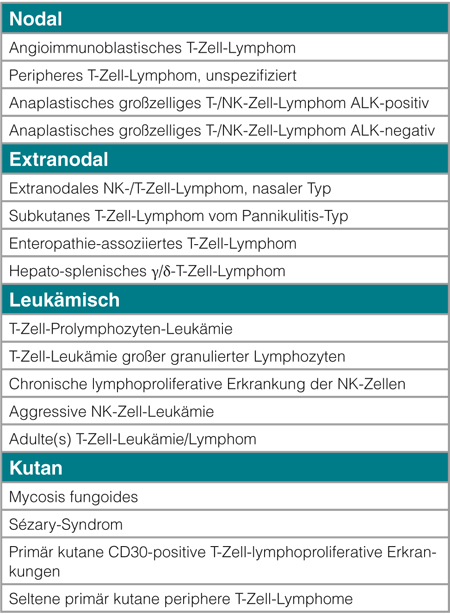
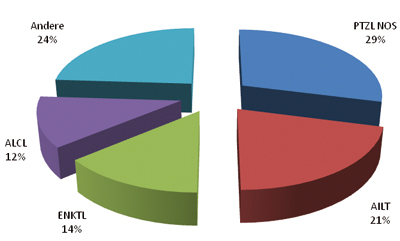
Die sehr heterogene Gruppe maligner (Non-Hodgkin-)Lymphome (NHL), die sich von T-Lymphozyten bzw. ihren Vorläufern sowie von Natürlichen Killer-Zellen (NK-Zellen) ableiten, werden als T-/NK-Zell-Lymphome bezeichnet. In der aktuellen WHO-Klassifikation werden 18 verschiedene T-/NK-Zell-Lymphome unterschieden. Eine Sonderform stellt das/die unreife (Vorläufer-)T-Zell-Lymphom/Leukämie dar, eine Entität, die biologisch, klinisch und therapeutisch in die Gruppe der akuten lymphatischen Leukämien zu zählen ist und im Folgenden nicht weiter besprochen wird. Die übrigen reifen sog. peripheren T-/NK-Zell-Lymphome (PTZL) machen weltweit nur etwa 10% aller NHL aus [1]. Dabei bestehen jedoch erhebliche regionale Unterschiede. So finden sich insbesondere die Virus-assoziierten PTZL mit deutlich höherer Inzidenz in Ostasien und der Karibik [2, 3]. Anders als die B-Zell-Lymphome, die in indolente (low grade, früher: niedrig-maligne) und aggressive (high-grade, früher: hoch-maligne) Lymphome eingeteilt werden, orientiert sich die Klassifikation der PTZL an der Primärmanifestation der Erkrankung. Demzufolge werden in der WHO-Klassifikation nodale, extranodale, kutane und leukämisch verlaufende PTZL unterschieden (Tab. 1, [4]). Die häufigsten Entitäten sind das periphere T-Zell-Lymphom, unspezifiziert (not otherwise specified; PTZL NOS), das angioimmunoblastische T-Zell-Lymphom (AITL), das extranodale NK-/T-Zell-Lymphom, nasaler Typ (ENKTL) und das anaplastische großzellige T-/NK-Zell-Lymphom (ALCL) mit oder ohne Expression der anaplastischen Lymphomkinase (ALK). Diese Entitäten sind weltweit für etwa 70–80% aller PTZL verantwortlich (Abb. 1, [5]). Im Allgemeinen zeigen die nodalen und extranodalen PTZL einen aggressiven Verlauf. Demgegenüber verhalten sich die seltenen leukämischen sowie die kutanen PTZL in der Regel lange indolent und werden, einer engeren klinischen Definition folgend, von den übrigen PTZL abgegrenzt und in dieser Übersicht nicht näher behandelt.
Ätiologie und Pathogenese
Über die Ätiologie der meisten PTZL ist wenig bekannt. Virusinfektionen scheinen für die Pathogenese einiger Entitäten eine wesentliche Rolle zu spielen. So kann in der ganz überwiegenden Mehrzahl der Fälle beim ENKTL eine chronische Infektion der malignen Zellen durch das Epstein-Barr-Virus (EBV) und bei der adulten T-Zell-Leukämie das humane T-Zell-Leukämie-Virus Typ 1 (HTLV1) nachgewiesen werden. Die genaue Pathophysiologie, die zur malignen Entartung führt, ist insbesondere unter Berücksichtigung der Tatsache, dass angesichts des hohen Durchseuchungsgrades mit EBV die Inzidenz des ENKTL vergleichsweise niedrig ist, bislang weitgehend ungeklärt. Als weitere ätiologische Faktoren werden (Auto-)Immunprozesse diskutiert. So tritt das Enteropathie-assoziierte T-Zell-Lymphom nahezu ausschließlich bei Patienten mit einer Zöliakie auf, wobei letztere bei subklinischem Verlauf gelegentlich erst durch die Lymphom-Diagnose erkannt wird. Für das hepato-splenische T-Zell-Lymphom wird eine Häufung nach Organtransplantation beschrieben. Zudem können erworbene genetische Aberrationen bei einigen PTZL nachgewiesen werden. Häufigstes Beispiel ist das ALK-positive
ALCL, das meist eine Translokation t(2;5) aufweist. Das entstehende Fusionsprodukt führt zu einer Störung der Apoptose und zu gesteigertem Zellwachstum und ist somit als (mit-)ursächlich für die Entstehung dieses Lymphoms anzusehen.
Klinik und Diagnostik
Typische Symptome von PTZL gibt es nicht. Neben Allgemeinsymptomen wie Schwäche und Müdigkeit findet sich bei einem Teil der Patienten eine sogenannte B-Symptomatik (Fieber > 38 °C ohne sonstige Ursache, Gewichtsverlust von mindestens 10% des Körpergewichts in den letzten sechs Monaten und Nachtschweiß mit Wäschewechsel). Lymphknotenschwellungen treten überwiegend bei den primär nodal verlaufenden PTZL auf, insgesamt manifestieren sich die PTZL jedoch häufiger als die B-Zell-Lymphome extranodal. Gelegentlich lassen sich, insbesondere beim AITL, paraneoplastische Epiphänomene (positiver Rheumafaktor, Nachweis von Kälteagglutininen/Kryoglobulinen, polyklonale Gammopathie, Eosinophilie, Hämolyse-Zeichen) nachweisen, die an immunologische bzw. rheumatologische Erkrankungen denken lassen und erhebliche differenzialdiagnostische Schwierigkeiten bereiten können.
Zur sicheren Diagnosestellung ist die Beurteilung eines histologischen Präparates durch einen in der Lymphom-Diagnostik erfahrenen Pathologen erforderlich. Eine zytologische Untersuchung zum Beispiel nach einer Feinnadelaspiration reicht nicht aus. Anders als bei den meisten B-Zell-Lymphomen finden sich bei den PTZL häufig keine charakteristischen immunphänotypischen Marker. Daher wird neben der morphologischen Beurteilung und molekulargenetischen Untersuchungen der Nachweis eines monoklonal rearrangierten T-Zell-Rezeptor-Gens mittels Polymerasekettenreaktion (PCR) zur Diagnosestellung gefordert.
Die Stadieneinteilung der nodalen und extranodalen PTZL erfolgt nach der Ann Arbor-Klassifikation.
Prognose
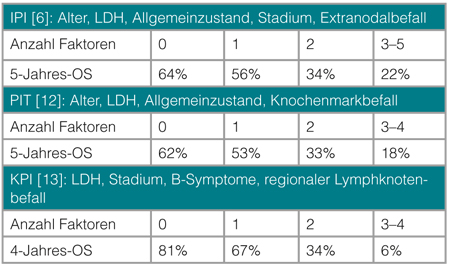
Die PTZL weisen insgesamt eine schlechte Prognose auf. Die Erkrankung verläuft meist entweder primär refraktär oder die Patienten erleiden ein Rezidiv. In der überwiegenden Zahl der Untersuchungen stellte der T-Zell-Phänotyp einen negativen prognostischen Faktor dar [6]. So liegt das mediane Gesamtüberleben bei etwa 9–42 Monaten [7–9]. Eine Ausnahme ist das ALK-positive ALCL. Bei dieser Entität können mit einer konventionellen Anthrazyklin-basierten (CHOP: Cyclophosphamid, Doxorubicin, Vincristin, Prednison) oder CHOP-ähnlichen Polychemotherapie Langzeitremissionen von über 70% erreicht werden [10, 11]. Mittlerweile wurden mehrere prognostische Indizes für PTZL publiziert. Die wichtigsten sind der bei aggressiven B-NHL etablierte Internationale Prognoseindex (IPI), der auch Gültigkeit bei PTZL besitzt [6]. Zudem haben der Prognoseindex für T-Zell-Lymphome (PIT, [12]) sowie für ENKTL der Korean Prognostic Index Bedeutung ([13], Tab. 2).
Therapie
Primärtherapie
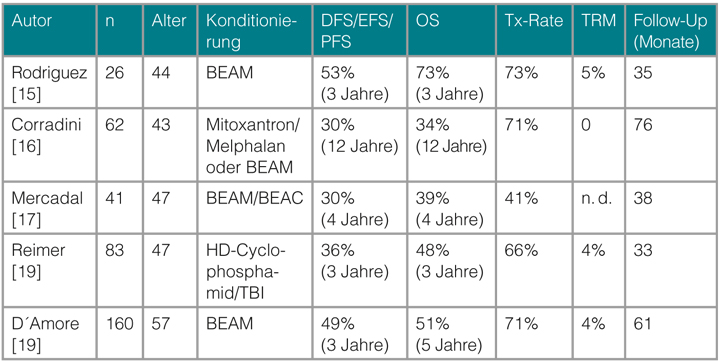
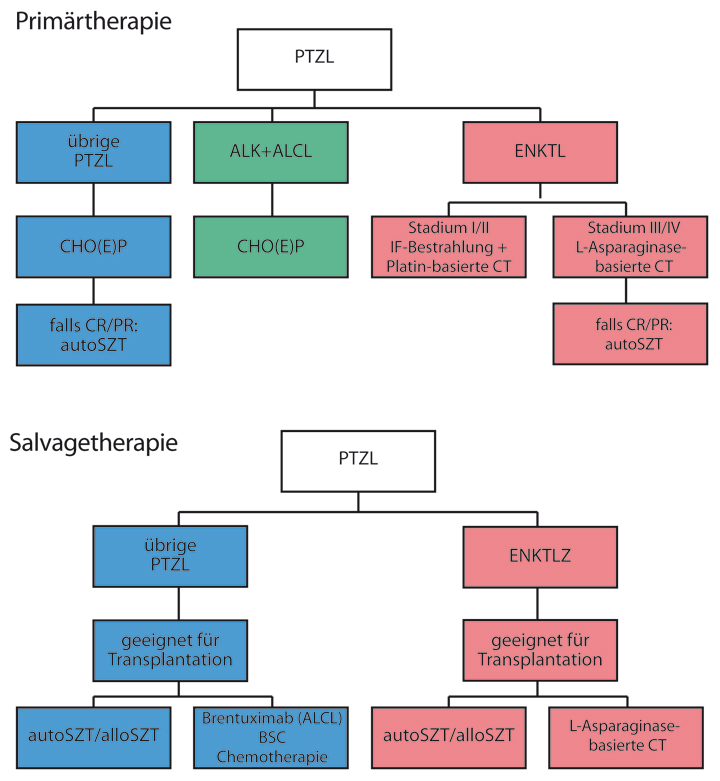
Eine Standardtherapie für PTZL ist nicht etabliert. Randomisierte Studien sind bislang nicht publiziert worden, in erster Linie wegen der Seltenheit der Erkrankungen. In Analogie zu den aggressiven B-NHL wird in der Erstlinienbehandlung meist eine Anthrazyklin-basierte (CHOP oder CHOP-ähnliche) Polychemotherapie bevorzugt. Abgesehen vom ALK-positiven ALCL werden mit dieser konventionellen Chemotherapie allerdings nur bei der Minderheit der Patienten langfristige Remissionen erreicht. Um die unbefriedigenden Ergebnisse in der Behandlung der PTZL zu verbessern, wird in vielen Zentren im Anschluss an eine Induktionstherapie eine Konsolidierung mittels Hochdosischemotherapie und autologer Blutstammzelltransplantation (autoSZT) durchgeführt. Der Wert der autoSZT ist bislang allerdings nicht durch randomisierte Studien eindeutig belegt. Fünf prospektive einarmige Studien haben diesen Therapieansatz untersucht [14–18]. In den beiden größten Serien mit 160 bzw. 83 Patienten lagen die Überlebensraten bei 51% nach fünf Jahren bzw. 48% nach drei Jahren [17, 18]. Die Studienergebnisse zeigten übereinstimmend, dass Patienten mit Chemotherapie-sensitiver Erkrankung, die eine gute Remission durch die Induktionstherapie erreichten, von dieser Therapieintensivierung profitierten, sodass dieses Vorgehen für diese Patienten einen sinnvollen Therapieansatz darstellt.
Eine Schwierigkeit in der Therapie der PTZL liegt im Risiko eines frühen Krankheitsprogresses, der dann auch Auswirkungen auf die Durchführbarkeit einer autoSZT hat. So lag in den genannten Studien die Transplantationsrate zwischen 41% und 73%. Um die Remissionsrate zu erhöhen, wurden verschiedene Ansätze untersucht. Für eine Intensivierung der Induktionschemotherapie sprechen Daten einer retrospektiven Subgruppenanalyse der Deutschen Studiengruppe für Hochmaligne Non-Hodgkin-Lymphome (DSHNHL). Dabei fand sich eine Verbesserung des ereignisfreien Überlebens durch die Hinzunahme des Topoisomerase-II-Hemmers Etoposid, was allerdings nur in der Gruppe der ALK-positiven ALCL Signifikanzniveau erreichte [19].
Einen anderen Ansatz verfolgte die Nordic Lymphoma Study Group, die randomisiert den Einsatz des anti-CD52-Antikörper Alemtuzumab in Kombination mit einer CHOP-Therapie als Induktionsbehandlung vor geplanter Hochdosistherapie bei jüngeren Patienten untersucht hat. Ergebnisse dieser Studie sind frühestens 2016 zu erwarten. Tab. 3 fasst die Ergebnisse der prospektiven Studien zur primären autoSZT zusammen. Ob die Ergebnisse der Erstlinienbehandlung durch eine allogene SZT (alloSZT) verbessert werden können, wird durch eine multinationale randomisierte Phase-III-Studie untersucht, die den Einsatz einer autoSZT mit dem einer alloSZT vergleicht. Ergebnisse dieser Studie stehen allerdings noch aus.
Abweichend von den übrigen PTZL stellt die (Involved-Field)-Bestrahlung einen wesentlichen Teil der Behandlung in den lokalisierten Ann Arbor-Stadien I und II des ENKTL dar. Eine multimodale Radiochemotherapie zeigte sich in retrospektiven Untersuchungen der alleinigen Chemotherapie oder Bestrahlung überlegen, wobei wegen der Gefahr eines raschen lokalen Krankheitsprogresses die Bestrahlung früh in das multimodale Behandlungskonzept integriert werden sollte [20]. Da ENKTL meist P-Glykoprotein in hoher Konzentration exprimieren, welches eine Multidrug-Resistenz (MDR) vermittelt, sind Anthrazyklin-basierte Chemotherapien (wie z. B. CHOP) nicht wirksam. In den letzten Jahren konnten überzeugende Daten zu Platin-haltigen Polychemotherapien publiziert werden. In den fortgeschrittenen Krankheitsstadien kann eine L-Asparaginase-haltige Polychemotherapie (z. B. nach dem SMILE-Protokoll: Dexamethason, Methotrexat, Ifosfamid, L-Asparaginase, Etoposid) mittlerweile als überlegene Systemtherapie angesehen werden [21], auch wenn randomisierte Untersuchungen fehlen. Allerdings sind vergleichbare Ansprechraten und Überlebensdaten durch andere Substanzkombinationen bislang nicht erreicht worden.
Salvagetherapie
Die meisten Patienten mit PTZL erleiden früher oder später einen Krankheitsprogress oder ein Rezidiv ihrer Erkrankung. Ebenso wie in der Primärtherapie ist ein Standardvorgehen in der Zweitlinientherapie bislang nicht etabliert. Eine kürzlich publizierte retrospektive Studie konnte zeigen, dass eine konventionell dosierte Chemotherapie in der Rezidivsituation keinen relevanten Einfluss mehr auf den Erkrankungsverlauf hat und einer rein supportiven Therapie nicht überlegen ist [22]. Eine Ausnahme dürfte das ENKTL darstellen, bei dem wie oben erwähnt auch im Rezidiv mit einer L-Asparaginase-haltigen Polychemotherapie häufig noch gute Ergebnisse erzielt werden können [21].
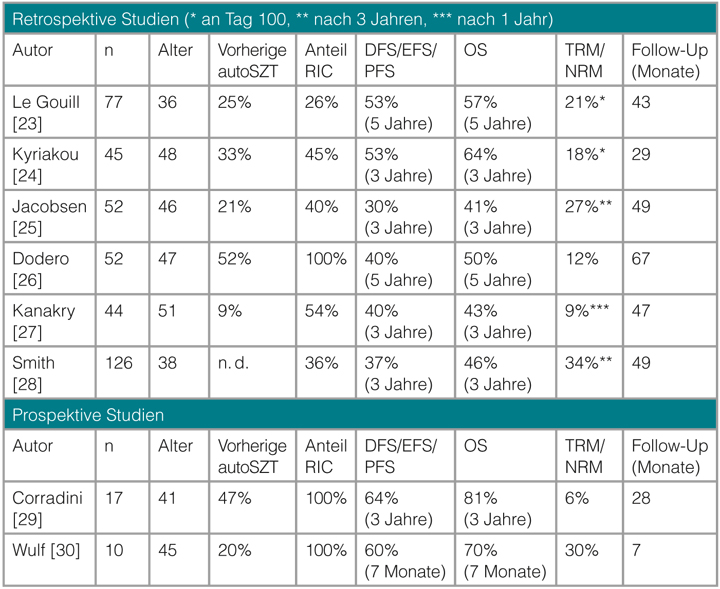
Sofern in der Erstlinientherapie keine autoSZT als Konsolidierungstherapie durchgeführt wurde, ist die Hochdosischemotherapie mit autoSZT eine mögliche Therapieoption im Rezidiv. Für Patienten mit PTZL wurde in dieser Situation in älteren vergleichenden Untersuchungen ein Ansprechen berichtet, das dem bei aggressiven B-NHL nicht unterlegen scheint. Allerdings unterliegen diese Arbeiten durch ihren retrospektiven Charakter einem Bias. Ein prinzipiell kurativer Ansatz kann mit einer alloSZT verfolgt werden. Die meisten retrospektiven Studien zeigen ein Plateau der Überlebenskurven [23–28]. Dieses ist möglicherweise (mit-)begründet durch einen für diesen Behandlungsansatz nachgewiesenen Graft-versus-Lymphoma-Effekt. Die größten retrospektiven Untersuchungen zur alloSZT wurde vom CIBMTR (Center for International Blood and Marrow Transplant Research) 2013 und der Société Française de Greffe de Moëlle et de Thérapie Cellulaire (SFGM-TC) publiziert. In der amerikanischen Studie wurden 126 Patienten überwiegend mit dosisreduzierter Konditionierung allogen transplantiert, wobei das Gesamtüberleben (OS) und das progressionsfreie Überleben (PFS) nach drei Jahren bei 42% bzw. 31% lagen [28]. Die nicht Rezidiv-bedingte Mortalität lag bei 34%. In der französischen Serie mit 77 Patienten fand sich nach überwiegend myeloablativer Konditionierung ein 5-Jahres-OS und -PFS von 57% bzw. 53% bei einer Therapie-assoziierten Mortalität (TRM) von 33% [23]. In beiden Studien konnte im Langzeitverlauf ein Plateau der Überlebenskurven erreicht werden, was für das kurative Potenzial zumindest in einer Subgruppe von Patienten spricht. In der größeren der beiden prospektiven Serien zur allogenen Transplantation bei rezidivierten oder refraktären PTZL konnte mit einem dosisreduzierten Konditionierungsprotokoll nach einer medianen Nachbeobachtung von 28 Monaten bei 71% der Patienten eine komplette Remission erzielt werden [29, 30]. Corradini et al. fanden bei
17 Patienten, von denen acht bereits eine autoSZT erhalten hatten, ein 3-Jahres-OS/-PFS von 81% bzw. 64%. Die TRM lag bei lediglich 6%.
Tab. 4 zeigt die Daten zur alloSZT in der Salvagetherapie der PTZL.
Neue Substanzen
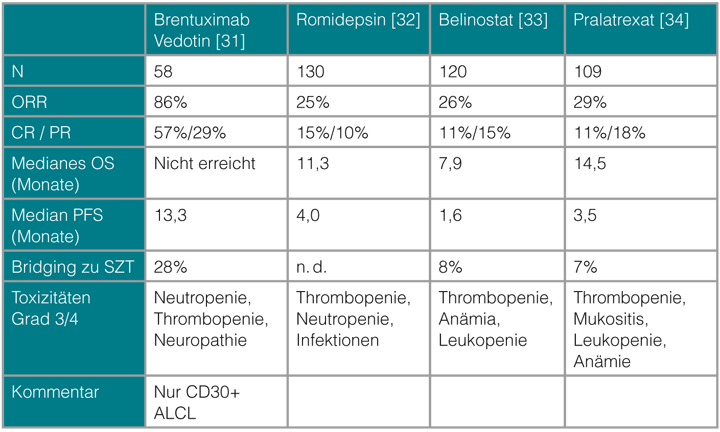
Eine große Anzahl neuer, z. T. zielgerichteter Substanzen ist in den letzten Jahren in der Behandlung der PTZL untersucht worden. Am überzeugendsten sind die Ergebnisse für das Antikörper-Wirkstoff-Konjugat Brentuximab Vedotin. Der chimäre anti-CD30-Antikörper Brentuximab ist mit dem Spindelgift Monomethyl-Auristatin E gekoppelt und entfaltet seine Wirkung über die spezifische Bindung an das vor allem auf ALCL unabhängig vom ALK-Status exprimierte CD30-Antigen. In einer prospektiven einarmigen Phase-II-Studie mit 58 vorbehandelten Patienten mit CD30-positiven ALCL konnte ein Gesamtansprechen von 86% mit einer Komplettremissionsrate von 57% und einem medianen PFS von
13,3 Monaten erzielt werden [31]. Hauptnebenwirkungen sind eine sensorische Polyneuropathie und eine Myelosuppression. Seit 2012 ist Brentuximab Vedotin durch die EMA (European Medicines Agency) zur Behandlung rezidivierter und refraktärer ALCL zugelassen und damit in Deutschland die erste und bislang einzige zugelassene Substanz für PTZL.
Eine weitere Klasse von Medikamenten, die eine Wirksamkeit bei PTZL gezeigt hat, sind die Histondeacetylase-Inhibitoren (HDACi). Mit dem selektiven Klasse-I-HDACi Romidepsin konnte in einer Phase-II-Studie mit
130 Patienten in der Salvage-Therapie eine Gesamtansprechrate von 25% (15% CR) erreicht werden. Die mediane Dauer des Ansprechens lag bei 17 Monaten. Die Nebenwirkungen waren moderat und betrafen in erster Linie Thrombozytopenie, Neutropenie und Infektionen [32]. Mit dem pan-HDACi Belinostat konnten bei 120 Patienten mit refraktären und rezidivierten PTZL ähnliche Ergebnisse erzielt werden (ORR 26%, CR 11%). Auch hier waren die Nebenwirkungen gering und betrafen vor allem die Hämatopoese [33]. Beide Substanzen werden wegen ihres günstigen Nebenwirkungsprofils in aktuell laufenden Studien in Kombination mit einer Chemotherapie untersucht.
Pralatrexat ist ein dem Methotrexat verwandter Folsäure-Antagonist, der wegen seiner stärkeren Aufnahme in die Zelle und der längeren intrazellulären Retention effektiver wirkt. Bei den
PTZL konnte in der Salvage-Therapie in einer einarmigen Studie eine Ansprechrate von 29% (CR 11%) bei 109 Patienten erreicht werden. Dabei waren die wesentlichen Nebenwirkungen Mukositis und Myelosuppression [34]. Romidepsin, Belinostat und Pralatrexat sind für die Rezidivtherapie der PTZL bislang nur in den USA zugelassen. Tab. 5 fasst die wichtigsten Studienergebnisse mit den neuen Substanzen zusammen.
Zahlreiche weitere Medikamente (Antikörper, Aurorakinase-Inhibitoren, ALK-Inhibitoren etc.) werden gegenwärtig in Studien untersucht.
Einen möglichen Therapiealgorithmus für die Primär- und Salvage-Therapie der PTZL zeigt Abb. 2.
Literatur
1. Anonymous. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. The Non-Hodgkin's Lymphoma Classification Project. Blood 1997; 89: 3909-18.
2. Nakamura S et al. Clinicopathologic study of 212 cases of peripheral T-cell lymphoma among the Japanese. Cancer 1993; 72: 1762-72.
3. Swerdlow SH et al. Caribbean T-cell lymphoma/leukemia. Cancer 1984; 54: 687-96.
4. Catovsky D et al. Chapter 11. In: Swerdlow H, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition. IARC Press, Lyon 2008: 269-319.
5. Vose J, Armitage J, Weisenburger D; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J Clin Oncol 2008; 26: 4124-30.
6. Gisselbrecht C et al. Prognostic significance of T-cell phenotype in aggressive non-Hodgkin's lymphomas. Groupe d'Etudes des Lymphomes de l'Adulte (GELA). Blood 1998; 92: 76-82.
7. Huang HQ et al. Clinical outcomes of 106 patients with peripheral T-cell lymphoma treated by standard CHOP regimen. Ai Zheng 2004; 23 (Suppl): 1443-7.
8. Kim K et al. Clinical features of peripheral T-cell lymphomas in 78 patients diagnosed according to the Revised European-American lymphoma (REAL) classification. Eur J Cancer 2002; 38: 75-81.
9. López-Guillermo AI et al. Peripheral T-cell lymphomas: Initial features, natural history, and prognostic factors in a series of 174 patients diagnosed according to the R.E.A.L. Classification. Ann Oncol 1998; 9: 849-55.
10. Gascoyne RD D et al. Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood 1999; 93: 3913-21.
11. Falini B et al. ALK+ lymphoma: Clinico-pathological findings and outcome. Blood 1999; 93: 2697-706.
12. Gallamini A et al. Peripheral T-cell lymphoma unspecified (PTCL-U): A new prognostic model from a retrospective multicentric clinical study. Blood 2004; 103: 2474-9.
13. Lee J et al. Extranodal natural killer T-cell lymphoma, nasal-type: A prognostic model from a retrospective multicenter study. J Clin Oncol 2006; 24: 612-8.
14. Corradini P et al. Long-term follow-up of patients with peripheral T-cell lymphomas treated up-front with high-dose chemotherapy followed by autologous stem cell transplantation. Leukemia 2006; 20: 1533-8.
15. Rodríguez J et al. Frontline autologous stem cell transplantation in high-risk peripheral T-cell lymphoma: A prospective study from The Gel-Tamo Study Group. Eur J Haematol 2007; 79: 32-8.
16. Mercadal S et al. Intensive chemotherapy (high-dose CHOP/ESHAP regimen) followed by autologous stem-cell transplantation in previously untreated patients with peripheral T-cell lymphoma. Ann Oncol 2008; 19: 958-63.
17. Reimer P et al. Autologous stem cell transplantation (autoSCT) as first-line therapy in peripheral T cell lymphomas (PTCL). Results of a prospective multicenter study. J Clin Oncol 2009; 27: 106-13.
18. D´Amore F et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol 2012; 30: 3093-9.
19. Schmitz N et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma: An analysis of patients with T-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood 2010; 116: 3418-25.
20. Kwong YL, Anderson BO, Advani R et al; Asian Oncology Summit. Management of T-cell and natural-killer-cell neoplasms in Asia: Consensus statement from the Asian Oncology Summit 2009. Lancet Oncol 2009; 10: 1093-101.
21. Yamaguchi M et al. Phase II study of SMILE chemotherapy for newly diagnosed stage IV, relapsed, or refractory extranodal natural killer (NK)/T-cell lymphoma, nasal type: The NK-Cell Tumor Study Group study. J Clin Oncol 2011; 29: 4410-6.
22. Mak V et al. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: Spectrum of disease and rare long-term survivors. J Clin Oncol 2013; 31, 1970-6.
23. Le Gouill S et al. Graft-versus-lymphoma effect for aggressive T-cell lymphoma in adults: A study by the Société Française de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 2008; 26: 2264-71.
24. Kyriakou C et al. Allogeneic stem cell transplantation is able to induce long-term remissions in angioimmunoblastic T-cell lymphoma: A retrospective study from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2009; 27: 3951-8.
25. Jacobsen ED et al. A large single-center experience with allogeneic stem-cell transplantation for peripheral T-cell non-Hodgkin lymphoma and advanced mycosis fungoides/Sezary syndrome. Ann Oncol 2011; 22: 1608-13.
26. Dodero A et al. Allogeneic transplantation following a reduced-intensity conditioning regimen in relapsed/refractory peripheral T-cell lymphomas: Long-term remissions and response to donor lymphocyte infusions support the role of a graft-versus-lymphoma effect. Leukemia 2012; 26: 520-6.
27. Kanakry JA et al. Outcomes of related donor HLA-identical or HLA-haploidentical allogeneic blood or marrow transplantation for peripheral T cell lymphoma. Biol Blood Marrow Transplant 2013; 19: 602-6.
28. Smith SM et al. Hematopoietic cell transplantation for systemic mature T-cell non-Hodgkin lymphoma. J Clin Oncol. 2013; 31: 3100-9.
29. Corradini P et al. Graft-versus-lymphoma effect in relapsed peripheral T-cell non-Hodgkin's lymphomas after reduced-intensity conditioning followed by allogeneic transplantation for hematopoietic cells. J Clin Oncol 2004; 22: 2172-6.
30. Wulf GG et al. Reduced intensity conditioning and allogeneic stem cell transplantation after salvage therapy integrating alemtuzumab for patients with relapsed peripheral T-cell non-Hodgkin's lymphoma. Bone Marrow Transplant 2005; 36: 271-3.
31. Pro B et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J Clin Oncol 2012; 30: 2190-6.
32. Coiffier B et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 2012; 30: 631-6.
33. O'Connor OA et al. Belinostat, a novel pan-histone deacetylase inhibitor (HDACi), in relapsed or refractory peripheral T-cell lymphoma (R/R PTCL): Results from the BELIEF trial. J Clin Oncol 2013; 31 (15S): 519s (ASCO 2013, Abstract #8507).
34. O'Connor OA et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: Results from the pivotal PROPEL study. J Clin Oncol 2011; 29: 1182-9.

Prof. Dr. med. Peter Reimer
Klinik für Hämatologie, Internistische
Onkologie und Stammzelltransplantation
Evangelisches Krankenhaus
Essen-Werden gGmbH
Pattbergstr. 1-3, 45239 Essen
+49 201 4089-2230
+49 201 4089-2297
p.reimer[at]kliniken-essen-sued[dot]de