Aktuelle Therapiestandards und neue therapeutische Strategien beim Mantelzell-Lymphom
Andrea Schnaiter, Martin Dreyling
Noch immer gibt es keinen kurativen Ansatz zur Behandlung des Mantelzell-Lymphoms, die Wahl der Therapie richtet sich vielmehr nach Alter und Allgemeinzustand des Patienten sowie nach dem Ausbreitungsstadium. In den letzten Jahren haben sich jedoch zahlreiche erfolgversprechende Therapieoptionen aufgetan. Bei der Behandlung jüngerer Patienten in gutem Allgemeinzustand wurde hochdosiertes Cytarabin fester Bestandteil der Erstlinientherapie. Durch den CD20-Antikörper Rituximab konnten die Überlebensraten deutlich verbessert werden. Der Proteasomeninhibitor Bortezomib stellt in Kombination mit konventioneller Immunchemotherapie eine zugelassene Alternative für Patienten dar, die nicht für eine Hochdosistherapie geeignet sind. Für die refraktäre und/oder rezidivierte Situation stehen weitere zielgerichtete Nicht-Chemotherapeutika wie Ibrutinib oder Temsirolimus, möglicherweise bald auch Lenalidomid zur Verfügung.
Klinik
In den meisten Fällen liegt bereits bei Erstdiagnose eines Mantelzell-Lymphoms ein Stadium III oder IV nach Ann Arbor vor. Klinische Charakteristika sind Lymphadenopathie, Hepato- und Splenomegalie und Knochenmarkinfiltration. Daneben finden sich bei fast allen Patienten in der Durchflusszytometrie Lymphomzellen im peripheren Blut [1]. Sehr häufig manifestiert sich das Mantelzell-Lymphom auch im Gastrointestinaltrakt. Besonders in den frühen Stadien I und II nach Ann Arbor wird deshalb im Rahmen der Ausbreitungsdiagnostik auch die Durchführung einer endoskopischen Diagnostik empfohlen [2].
Diagnose
Entscheidend für die Diagnosestellung ist das Vorhandensein der Translokation t(11;14) in der Fluoreszenz-In-Situ-Hybridisierung (FISH) sowie die Überexpression von Cyclin D1 in der Immunhistochemie. Hier finden sich außerdem in der Regel eine Koexpression von CD5 und B-Zell-Antigenen, sowie eine Expression von BCL2. Im Unterschied zur chronischen lymphatischen Leukämie (CLL) ist die Expression von CD23 nur schwach positiv oder fehlend.
Der IGHV-Status ist in der Regel unmutiert, passend zu einer B-Zelle vor dem Transit durch das Keimzentrum. Im Gegensatz zur CLL korreliert der IGHV-Status beim Mantelzell-Lymphom nicht mit dem Überleben oder der ZAP70-Expression.
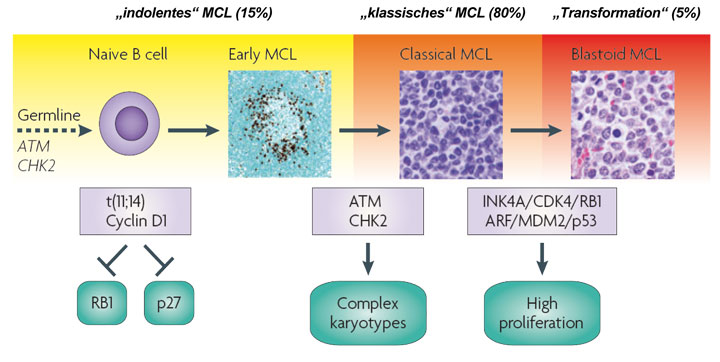
Es existieren unterschiedliche morphologische Varianten, sodass die Unterscheidung von Marginalzonen-Lymphomen, CLL/SLL oder bei leukämischem Verlauf auch von akuten Leukämien nicht immer leicht fällt. Klinisch und prognostisch besonders relevant sind wegen ihres aggressiven Verlaufs die blastoiden und pleomorphen Varianten des Mantelzell-Lymphoms. Der Anteil Ki67-positiver Zellkerne hat hohe prognostische Bedeutung und ist der bisher am besten etablierte histomorphologische Risikofaktor [3].
Zur klinischen Abschätzung der Prognose dient der Mantle Cell Lymphoma International Prognostic Index (MIPI). In diesen Prognoseindex fließen das Patientenalter, der ECOG-Status, die Konzentration der Laktatdehydrogenase (LDH) und die Leukozytenzahl ein.
Pathogenese
Die Translokation t(11;14) gilt als primäres genetisches Ereignis bei der Entstehung des Mantelzell-Lymphoms. Sie tritt in der unreifen B-Zelle auf und führt zu einer konstitutiven Überexpression von Cyclin D1. Dadurch kommt es letztendlich zur Aktivierung des Zellzyklus mit Übertritt der Zelle von der G1- in die S-Phase und damit zur Zellproliferation.
Beim Mantelzell-Lymphom sind sowohl der normale Zellzyklus als auch DNA-Reparaturwege gestört. Ursache sind eine Überexpression von Cyclin D1, ein Verlust des Inhibitors des Cyclin D1/CDK4/6-Komplexes p16INK4a, Deletionen von RB, gesteigerte Expression von CDK4 und BMI1 und eine Deregulierung von E2F. Häufig sind auch Mutationen oder Deletionen von ATM und TP53 sowie die Überexpression von MDM2 [4].
Aktuelle Therapiestandards
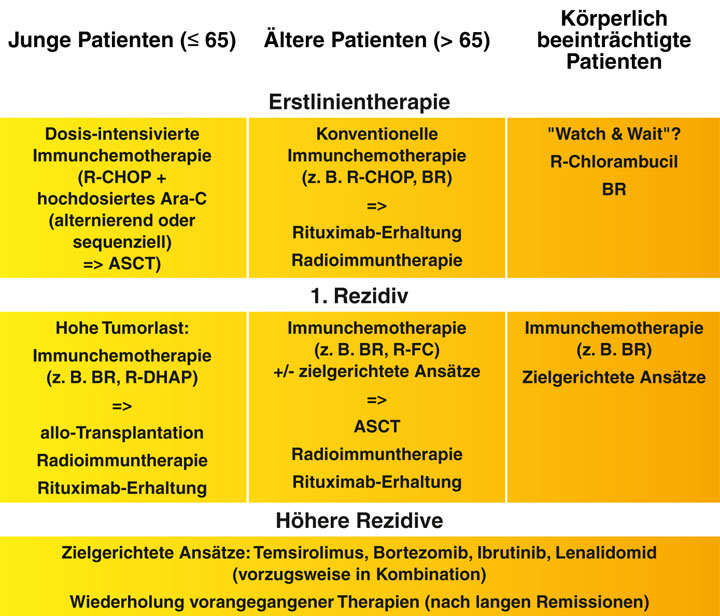
Die Therapie des Mantelzell-Lymphoms richtet sich nach dem Patientenalter, insbesondere aber auch nach dem körperlichen Zustand des Patienten. Auch das Ausbreitungsstadium hat Einfluss auf die Wahl der Therapie.
Eine Therapieindikation besteht in der Regel direkt nach Diagnosestellung bei symptomatischer Erkrankung. Nur bei 10–15% der Patienten liegt ein eher indolenter Subtyp des Mantelzell-Lymphoms vor. Eine klare Vorhersage zum Verlauf kann bisher nicht gegeben werden. Patienten mit indolentem Verlauf präsentieren sich in der Regel jedoch in gutem Allgemeinzustand und haben normale LDH-Werte. Häufig besteht keine Lymphadenopathie, es finden sich jedoch Splenomegalie, Knochenmarkinfiltration und Lymphomzellen im peripheren Blut. Weitere Hinweise auf einen indolenten Verlauf können ein niedriger Ki67-Anteil von < 10% oder Negativität für SOX-11 sein.
In diesen Fällen kann unter engmaschiger Überwachung eine "Watch & Wait"-Strategie verfolgt werden, wie eine retrospektive Analyse der Daten von 97 Patienten zeigt. 32% der Patienten wurden länger als drei Monate vor Therapieeinleitung beobachtet, drei Patienten wurden sogar länger als fünf Jahre beobachtet. Die mediane Zeit bis zur Therapieeinleitung betrug immerhin zwölf Monate. Patienten, die nicht direkt nach Diagnosestellung therapiert wurden, hatten häufiger einen besseren Performance Status und einen niedrigeren MIPI. Die Beobachtungsgruppe schnitt auch bezüglich des Gesamtüberlebens besser ab als die Gruppe der Patienten, bei der frühzeitig eine Therapie eingeleitet werden musste. Ein Nachteil im Gesamtüberleben im Vergleich zu unmittelbar therapierten Patienten ergab sich nicht [5].
Ältere Patienten
Das mediane Alter bei Diagnosestellung liegt beim Mantelzell-Lymphoms bei ca. 65 Jahren. Die Gruppe der älteren Patienten ist sehr heterogen, was die körperliche Leistungsfähigkeit und die Organfunktionen angeht. Für die Wahl der geeigneten Therapie spielt hier nicht das chronologische, sondern das biologische Alter eine große Rolle. In der Erstlinientherapie fitter Patienten im Alter von > 65 Jahren sollte eine konventionelle Immunchemotherapie mit Rituximab-Erhaltungstherapie eingesetzt werden. Therapieziel ist das Erreichen einer langfristigen Remission. Hierfür infrage kommende Schemata sind vorrangig Rituximab, Cyclophosphamid, Doxorubicin, Vincristin und Prednisolon (R-CHOP) oder Rituximab und Bendamustin (BR), alternativ bei sehr aggressivem Bild auch Rituximab, Bendamustin und Cytarabin (R-BAC).
Eine Studie des Europäischen MCL-Netzwerks (MCL elderly) an 560 Patienten im medianen Alter von 70 Jahren, die für eine Hochdosistherapie nicht geeignet waren, verglich randomisiert eine Erstlinientherapie mit Rituximab, Fludarabin und Cyclophosphamid (R-FC) mit dem R-CHOP-Schema. Bei Ansprechen wurden die Patienten einer Erhaltungstherapie mit Interferon α oder Rituximab zugeteilt. Die Behandlung mit R-FC hatte ein signifikant kürzeres Gesamtüberleben und häufiger hämatologische Toxizitäten zur Folge. R-CHOP erwies sich daher als geeignetere Therapieoption für diese Patientengruppe. Die Rituximab-Erhaltungstherapie konnte das Gesamtüberleben von Patienten, die auf R-CHOP ansprachen, nochmals deutlich verlängern (4-Jahres-Gesamtüberleben 87%, [6]).
Eine weitere randomisierte Multicenterstudie (NHL 1-2003) der Studiengruppe indolente Lymphome (StiL) prüfte die Kombination BR gegen R-CHOP in der Erstlinientherapie. 94 Patienten mit Mantelzell-Lymphom wurden eingeschlossen. Auch hier lag das mediane Alter der Patienten bei
70 Jahren. Es wurden in beiden Gruppen sehr gute Gesamtansprechraten erreicht (BR 93%, R-CHOP 91%). Das progressionsfreie Überleben in der mit BR behandelten Gruppe war sogar etwas länger als bei R-CHOP, das Gesamtüberleben war dagegen in beiden Gruppen vergleichbar. Insbesondere war aber die Verträglichkeit der Therapie mit BR besser (schwere unerwünschte Nebenwirkungen (SAE): BR 19%, R-CHOP 29%, [7]). Besonders bei Patienten mit Komorbiditäten stellt daher die Kombination BR eine gute Therapiealternative zu R-CHOP dar.
Die Kombination R-BAC wurde in einer Phase-II-Studie als Erstlinientherapie und bei rezidivierter oder refraktärer Erkrankung eingesetzt. Die Gesamtansprechrate lag bei 100% mit 95% kompletten Remissionen bei zuvor unbehandelten Patienten. Das progressionsfreie Überleben nach zwei Jahren lag in der Erstlinie bei 95%. Allerdings war die Rate an Hämatotoxizitäten sehr hoch: So mussten zwei Drittel der Patienten Thrombozytentransfusionen erhalten [8]. Dieses Regime sollte daher lediglich bei sehr fitten Hochrisiko-Patienten (blastoide Variante, sehr hohe LDH) erwogen werden. Für kompromittierte Patienten stehen weniger toxische Therapien wie BR, Rituximab mit Cyclophosphamid, Vincristin und Prednisolon (R-CVP) oder Rituximab mit Chlorambucil (R-Clb) zur Verfügung. Therapieziel ist hier die Krankheitskontrolle.
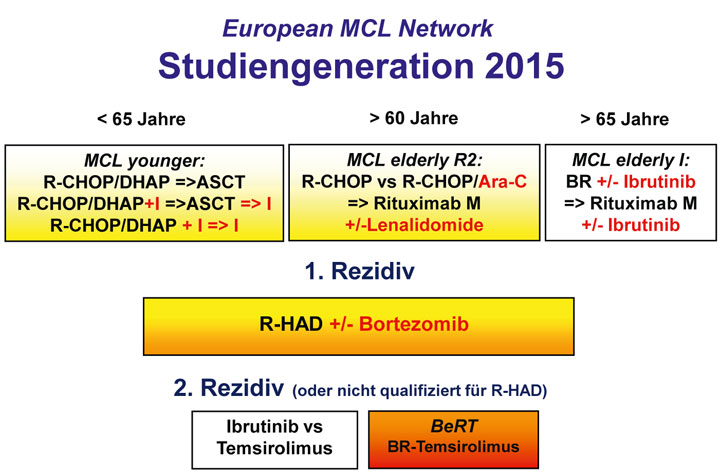
Da mit Cytarabin bei jüngeren Patienten sehr gute Ergebnisse erzielt wurden, prüft aktuell die MCL R2 elderly-Studie des Europäischen MCL-Netzwerks dieses Konzept (Abb. 3).
Jüngere Patienten
Therapiestandard in der Erstlinientherapie bei jüngeren Patienten in gutem Allgemeinzustand stellt heute eine Immunchemotherapie mit Rituximab und Hochdosis-Cytarabin sowie konsolidierender autologer Stammzelltransplantation dar [2]. Diese Empfehlung beruht auf einer Studie der Deutschen Studiengruppe Niedrigmaligne Lymphome (GLSG), in der Patienten im Alter von bis zu 65 Jahren eine Konsolidierung mit myeloablativer Radiochemotherapie oder Interferon α-Erhaltungstherapie erhielten. Im Transplantationsarm war das mediane progressionsfreie Überleben und in einer Metaanalyse sogar das Gesamtüberleben signifikant verlängert [9].
Vor Konsolidierung mittels autologer Stammzelltransplantation sollte eine Induktionstherapie mit hochdosiertem Cytarabin durchgeführt werden. In der MCL younger-Studie des Europäischen MCL-Netzwerks wurde eine alternierende Induktionstherapie mit R-CHOP und R-DHAP mit einer alleinigen R-CHOP-Therapie, jeweils gefolgt von einer myeloablativen Konsolidierung mit autologer Stammzelltransplantation, verglichen. Nach der Kombination mit Cytarabin wurde eine deutlich längere Zeit bis zum Therapieversagen (TTF: 88 vs. 46 Monate) und ein Trend zu einem längeren medianen Gesamtüberleben (noch nicht erreicht vs. 83 Monate) beobachtet [10], die Rate der molekularen Remissionen stieg von 32% auf 73% an [11]. Eine alleinige Hochdosis-Cytarabin-Therapie mit anschließender Hochdosis-Therapie und autologer Stammzelltransplantation ist dagegen nicht ausreichend wirksam [12].
Die LyMa Studie (Phase III) untersuchte den Stellenwert einer Rituximab-Erhaltungstherapie nach autologer Stammzelltransplantation. Eine erste Zwischenanalyse wurde auf dem ASH-Kongress 2014 präsentiert [13]. Alle Patienten erhielten vier Zyklen R-DHAP vor autologer Stammzelltransplantation, ggf. eine Salvagetherapie mit R-CHOP. Nach autologer Transplantation erhielten die Patienten eine Rituximab-Erhaltungstherapie über drei Jahre oder wurden lediglich beobachtet. Die erste Interimsanalyse weist auf ein verbessertes ereignisfreies und progressionsfreies Überleben nach Rituximab-Erhaltungstherapie hin. Wenn sich diese Tendenz auch in der Endauswertung bestätigt, wird eine solche Antikörper-Erhaltungstherapie auch bei jüngeren Patienten empfohlen.
Bei jüngeren Patienten mit Rezidiv nach autologer Transplantation oder refraktärer Erkrankung sollte eine allogene Stammzelltransplantation diskutiert werden; eine autologe Transplantation kommt nur infrage, wenn sie noch nicht in der Erstlinientherapie durchgeführt wurde. Eine retrospektive Analyse zur Konditionierung mit reduzierter Intensität (RIC) an 70 Patienten ergab ein 2-Jahres-Gesamtüberleben von 53%. Die Transplantations-assoziierte Mortalität nach zwei Jahren lag bei 32%. Der Remissionsstatus zum Zeitpunkt der Transplantation war der einzige Parameter, der das ereignisfreie und das Gesamtüberleben beeinflusste [14]. Eine experimentelle Option mit vielversprechenden Ergebnissen stellt bei jüngeren Patienten die haploidente Transplantation dar, wenn eine HLA-idente Transplantation mangels Spender nicht möglich ist [15].
Zielgerichtete Therapien
Im Rezidiv oder bei refraktärer Erkrankung kommen für Patienten, die nicht für eine allogene Transplantation geeignet sind, zielgerichtete Therapien in Kombination mit konventioneller Immunchemotherapie infrage. Sinnvoll ist die Behandlung im Rahmen einer klinischen Studie.
In den letzten Jahren haben solche zielgerichtete Substanzen, sogenannte „Small Molecules“, immer mehr an Stellenwert in der Therapie des Mantelzell-Lymphoms gewonnen. Viele dieser Substanzen weisen eine orale Bioverfügbarkeit und ein vorteilhaftes Verträglichkeitsprofil auf.
Molekulare Zielstrukturen beim Mantelzell-Lymphom sind u. a. das Proteasom und der B-Zell-Rezeptor-Signalweg mit mTOR, PI3K und AKT. Andere Angriffspunkte sind die Cyclin-abhängigen Kinasen oder die Apoptose-Regulation. Immunmodulatoren wie Lenalidomid führen zu einem Anstieg von NK-Zellen und peripheren regulatorischen T-Zellen (T-regs) und wirken u. a. über das Microenvironment.
Bortezomib
Das Proteasom kontrolliert die Stabilität vieler für den Zellzyklus bedeutsamer Proteine. Dazu gehören unter anderem Cycline, Cyclin-abhängige Kinasen (CDKs), verschiedene Tumorsuppressoren und NFkB. Proteasominhibitoren sensibilisieren Zellen für die Apoptose. Der Proteasominhibitor Bortezomib hemmt spezifisch diese Proteasomaktivität.
Seit Februar 2015 ist Bortezomib in Europa in Kombination mit Rituximab, Cyclophosphamid, Doxorubicin und Prednisolon (VR-CAP) für die Erstlinientherapie von Patienten mit Mantelzell-Lymphom, die für eine Stammzelltransplantation nicht geeignet sind, zugelassen. Grundlage dieser Zulassung sind Daten einer Phase-III-Studie, in der 487 Patienten 6–8 Zyklen R-CHOP oder eine ähnliche Chemotherapie mit Bortezomib (VR-CAP) erhielten [16]. Das mediane progressionsfreie Überleben lag bei 14,4 Monaten in der R-CHOP-Gruppe gegenüber 24,7 Monaten in der VR-CAP-Gruppe. Auch die Rate an kompletten Remissionen, die mediane Dauer der kompletten Remission, das mediane behandlungsfreie Intervall und das 4-Jahres-Gesamtüberleben waren im Bortezomib-Arm signifikant verbessert. Allerdings war die Rate an Grad-3/4-Thrombozytopenien in der VR-CAP-Gruppe deutlich höher, ein Viertel der Patienten erhielt Thrombozyten-Transfusionen.
Aktuell prüfen zahlreiche Studien Bortezomib in Kombination mit Chemotherapie (First-line: EPOCH-R, Rezidiv: R-HAD) oder molekularen Ansätzen (CDK4/6-Inhibitoren, Alisertib und Rituximab) im Rezidiv.
Temsirolimus
Da der mTOR-Signalweg beim Mantelzell-Lymphom konstitutiv aktiviert ist, stellt der spezifische mTOR-Inhibitor Temsirolimus eine besonders interessante Therapieoption dar: Temsirolimus führt zu einem Zellzyklus-Arrest in der G1-Phase und hemmt die Synthese von Cyclin D1.
Seit 2009 ist die Substanz für die Behandlung des rezidivierten und/oder refraktären Mantelzell-Lymphoms zugelassen. In der Zulassungsstudie wurden 162 Patienten mit rezidivierter oder refraktärer Erkrankung randomisiert zwischen zwei unterschiedlichen Dosisregimes von Temsirolimus oder einer Monochemotherapie nach Wahl des Prüfers: Eine dreiwöchige Induktion mit 175 mg Temsirolimus wöchentlich, gefolgt von einer Konsolidierung mit 75 mg pro Woche, verbesserte sowohl das progressionsfreie Überleben (175/75 mg: 4,8 Monate vs. „Investigator´s choice“: 1,9 Monate) als auch die Ansprechraten [17].
Eine Phase-I/II-Studie prüfte die Kombination von Temsirolimus mit BEndamustin-Rituximab im Rezidiv oder bei refraktärer Erkrankung (BERT-Studie). Im Phase-I-Teil mit
15 Patienten wurde keine dosislimitierende Toxizität beobachtet. Die Gesamtansprechrate lag bei 93% (14 von 15 Patienten), fünf Patienten erreichten eine komplette Remission (33%) bei einer progressionsfreien Überlebensrate von 67% nach 19 Monaten [18].
Lenalidomid
Der Immunmodulator Lenalidomid wurde im Jahr 2013 von der FDA für die Behandlung von Patienten mit rezidiviertem oder refraktärem Mantelzell-Lymphom zugelassen. Die Substanz ist oral bioverfügbar.
Eine Phase-II-Studie (NHL-003) untersuchte Lenalidomid bei rezidivierten oder therapierefraktären aggressiven Non-Hodgkin-Lymphomen als Monotherapie. Es wurden 25 mg täglich über 21 Tage in 28-Tages-Zyklen eingesetzt. Die Behandlung wurde bis zum Progress fortgesetzt oder bei Unverträglichkeit abgebrochen. Die Gesamtansprechrate betrug 42% (24 von 57 Patienten), das mediane progressionsfreie Überleben lag bei
5,7 Monaten, die häufigste unerwünschte Wirkung war eine Grad-4-Leukozytopenie [19].
Eine randomisierte Phase-II-Studie (MCL-002, Sprint) verglich darüber hinaus Lenalidomid mit einer Monochemotherapie beim rezidivierten Mantelzell-Lymphom. Lenalidomid wurde in einer täglichen Dosis von 25 mg über 21 Tage in 28-Tages-Zyklen eingesetzt. 254 Patienten wurden eingeschlossen, das mediane Alter lag bei 69 Jahren. Die Kohorte war stark vorbehandelt. Die Gesamtansprechrate im Lenalidomid-Arm lag bei 40% gegenüber 11% nach Chemotherapie (p = 0,001). Entsprechend wurde das mediane progressionsfreie Überleben von 5,7 auf 8,4 Monate verlängert (p = 0,004; [20]).
Die Ergebnisse einer Phase-I/II-Studie zur Kombination von Lenalidomid mit Rituximab in der refraktären oder rezidivierten Situation wurden 2012 publiziert: Die maximale tolerierte Dosis lag für Lenalidomid bei 20 mg, verabreicht an 21 Tagen eines 28-Tages-Zyklus. Für 44 Patienten der Phase II lag die Gesamtansprechrate bei 57% mit 36% kompletten Remissionen. Das mediane progressionsfreie Überleben lag bei 11,1 Monaten, das mediane Gesamtüberleben bei 24,3 Monaten [21].
Ibrutinib
Bruton’s Tyrosinkinase (BTK) spielt eine wichtige Rolle im B-Zell-Rezeptor-Signalweg. Der BTK-Inhibitor Ibrutinib wurde 2014 durch die EMA zur Behandlung des refraktären oder rezidivierten Mantelzell-Lymphoms zugelassen, basierend auf den Ergebnissen einer Phase-II-Studie, in der 111 Patienten mit refraktärer oder rezidivierter Erkrankung mit einer Dosis von 560 mg täglich behandelt worden waren. Das Nebenwirkungsprofil war günstig. Die Gesamtansprechrate lag bei 68% mit 21% kompletten und 47% partiellen Remissionen. Die mediane Ansprechdauer lag bei 17,5 Monaten, das geschätzte mediane progressionsfreie Überleben bei 13,9 Monaten. Das Gesamtüberleben nach 18 Monaten wurde auf 58% geschätzt [23].
Inzwischen gibt es allerdings Berichte zu frühen Ibrutinib-Resistenzen bei Patienten, die in einer Analyse des M. D. Anderson Cancer Center (n = 42) auf nachfolgende Therapien nicht mehr ansprachen und eine sehr schlechte Prognose hatten [24].
Ibrutinib wird aktuell in mehreren Studien in Kombination mit Immunchemotherapie-Regimes, aber auch mit anderen „Targeted Therapies“ wie Lenalidomid, PI3Kδ-Inhibitoren, CDK4/6-Inhibitoren und Bortezomib getestet.
Idelalisib
Idelalisib ist ein oral bioverfügbarer Inhibitor der Phosphoinositol-3-Kina-
se δ. Die Delta-Isoform ist für B-Lymphozyten besonders relevant. PI3Kδ führt durch Phosphorylierung zur Aktivierung von AKT und mTOR und spielt so eine wichtige Rolle in der Pathogenese lymphatischer Neoplasien; entsprechend führt die Inhibition der Kinase zum Zelltod.
Bisher ist die Substanz in Europa zur Behandlung der chronischen lymphatischen Leukämie und des follikulären Lymphoms zugelassen. Eine Phase-I-Studie mit 40 Patienten mit rezidiviertem Mantelzell-Lymphom zeigte eine Gesamtansprechrate von 40%, das mediane progressionsfreie Überleben lag jedoch nur bei 3,7 Monaten [25].
Aktive Studien beim Mantelzell-Lymphom laufen aktuell zu Kombinationen von Idelalisib mit verschiedenen CD20-Antikörpern, Immunmodulatoren und konventionellen Chemotherapeutika.
Literatur
1. Ferrer A et al. Leukemic involvement is a common feature in mantle cell lymphoma. Cancer 2007; 109: 2473-80.
2. Dreyling M et al. Newly diagnosed and relapsed mantle cell lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014; 25 (Suppl 3): iii83-92.
3. Determann O et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: Results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood 2008; 111: 2385-7.
4. Dreyling M on behalf of the European MCL Network. Mantle cell lymphoma: Biology, clinical presentation, and therapeutic approaches. American Society of Clinical Oncology 2014 Educational Book: 191-8.
5. Martin P et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009; 27: 1209-13.
6. Kluin-Nelemans HC et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012; 367: 520-31.
7. Rummel MJ et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: An open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013; 381: 1203-10.
8. Visco C et al. Combination of rituximab, bendamustine and cytarabine for patients with mantle cell non-Hodgkin lymphoma ineligible for intensive regimens or autologous transplantations. J Clin Oncol 2013; 31: 1442-9.
9. Lenz G et al. Myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission prolongs progression-free survival in follicular lymphoma: results of a prospective, randomized trial of the German Low-Grade Lymphoma Study Group. Blood 2004; 104: 2667-74.
10. Dreyling M et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: Results of a prospective randomized trial of the European MCL Network. Blood 2005; 105: 2677-84.
11. Hermine O et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose Ara-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) increases overall survival when compared to 6 courses of CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: Final analysis of the MCL younger trial of the European Mantle Cell Lymphoma Network (MCL net). Blood 2012; 120(21): ASH 2012, Abstract #151.
12. Metzner B et al. Long-term clinical and molecular remissions in patients with mantle cell lymphoma following high-dose therapy and autologous stem cell transplantation. Ann Hematol 2014; 93: 803-10.
13. Laurell A et al. High dose cytarabine with rituximab is not enough in first-line treatment of mantle cell lymphoma with high proliferation: Early closure of the Nordic Lymphoma Group Mantle Cell Lymphoma 5 trial. Leuk Lymphoma 2014; 55: 1206-8.
14. Le Gouill S et al. Rituximab maintenance versus wait and watch after four courses of R-DHAP followed by autologous stem cell transplantation in previously untreated young patients with mantle cell lymphoma: First interim analysis of the phase III prospective Lyma trial, a Lysa Study. Blood 2014; 124(21): ASH 2014, Abstract #146.
15. Le Gouill S et al. Reduced-intensity conditioning allogeneic stem cell transplantation for relapsed/refractory mantle cell lymphoma: A multicenter experience. Ann Oncol 2012; 23: 2695-703.
16. Zoellner AK et al. Sequential therapy combining clofarabine and T-cell-replete HLA-haploidentical haematopoietic SCT is feasible and shows efficacy in the treatment of refractory or relapsed aggressive lymphoma. Bone Marrow Transplant 2015; 50: 679-84.
17. Robak T et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 2015; 372: 944-53.
18. Hess G et al. Phase III study to evaluate temsirolimus compared with investigator's choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009; 27: 3822-9.
19. Hess G et al. Safety and efficacy of temsirolimus in combination with bendamustine and rituximab in relapsed mantle cell and follicular lymphoma. Leukemia 2015, Mar 13 [Epub ahead of print, DOI: 10.1038/leu.2015.60].
20. Witzig TE et al. An international phase II trial of single-agent lenalidomide for relapsed or refractory aggressive B-cell non-Hodgkin’s lymphoma. Ann Oncol 2011; 22: 1622-7.
21. Trneny M et al. Phase II randomized, multicenter study of lenalidomide vs best investigator’s choice in relapsed/refractory mantle cell lymphoma: Results of the MCL-002 (SPRINT) study. Blood 2014; 124(21): ASH 2014, Abstract #626.
22. Wang M et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: A phase 1/2 clinical trial. Lancet Oncol 2012; 13: 716-23.
23. Wang ML et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013; 369: 507-16.
24. Cheah CY et al. Patients with mantle cell lymphoma failing ibrutinib are unlikely to respond to salvage chemotherapy and have poor outcomes. Ann Oncol 2015, Feb 23 [Epub ahead of print, pii: mdv111].
25. Kahl BS et al. A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014; 123: 3398-405.

Dr. med. Andrea Schnaiter
Prof. Dr. med. Martin Dreyling
Klinikum der Universität München
Medizinische Klinik und Poliklinik III
Korrespondierender Autor:
Prof. Dr. med. Martin Dreyling
Koordinator des European Mantle-Cell
Lymphoma Network
Klinikum der Universität München
Medizinische Klinik und Poliklinik III
Marchioninistr. 15, 81337 München
+49 89 4400-72202
+49 89 4400-72201
martin.dreyling[at]med.uni-muenchen[dot]de