Neues zu Akuten Leukämien
ASH 2014
Die Prognose von Patienten mit akuten Leukämien wurde in jahrzehntelanger
Arbeit auch deutscher Studiengruppen schrittweise verbessert, aber die Situation ist vor allem bei erwachsenen Patienten nach wie vor unbefriedigend – abgesehen von wenigen Ausnahmen wie der akuten Promyelozyten-Leukämie (APL). Derzeit herrscht jedoch Aufbruchsstimmung auch auf diesem Gebiet, und das
war bei der Jahrestagung der American Society of Hematology (ASH) unübersehbar: Neben einer kaum überschaubaren Vielzahl von Kinaseinhibitoren und anderen „small molecules“ sind es vor allem immuntherapeutische Ansätze, die große Hoffnung machen.
Akute myeloische Leukämie (AML)
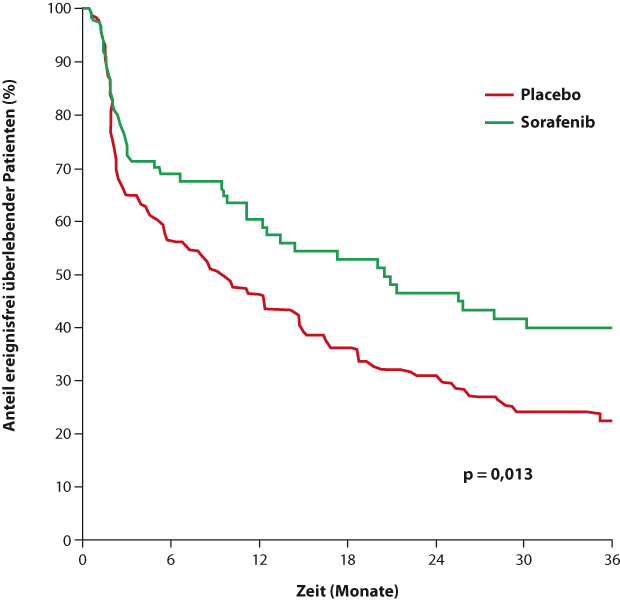
Tyrosinkinaseinhibitoren (TKI) haben in den letzten 15 Jahren die Onkologie verändert, angefangen bei der chronischen myeloischen Leukämie bis hin zur Behandlung verschiedener solider Tumoren. Sorafenib – bisher zur Therapie von Leber- und Nierenzellkarzinom zugelassen – hemmt eine ganze Reihe von Kinasen, und das macht die Substanz zu einem vielversprechenden Kandidaten für die AML, deren malignes Wachstum von zahlreichen verschiedenen Mutationen auch in Tyrosinkinasen angetrieben wird. Nach positiven Signalen aus nicht randomisierten Studien und Fallberichten schloss die deutsche Studienallianz Leukämien (SAL) 276 Patienten im Alter von bis zu 60 Jahren in ihre SORAML-Studie ein [1]. Sie erhielten eine Standard-Induktion mit Daunorubicin und Cytarabin und eine Konsolidierung mit hochdosiertem Cytarabin, dazu randomisiert und doppelblind Sorafenib oder Placebo, die hinterher auch noch für zwölf Monate als Erhaltungstherapie gegeben wurden. Patienten mit intermediärem und hohem Risiko sollten in der ersten Komplettremission allogen transplantiert werden.
Bei den Komplettremissionsraten, so Christop Röllig, Dresden, unterschieden sich beide Arme nicht (Sorafenib 60%, Placebo 59%), wohl aber beim primären Endpunkt ereignisfreies Überleben (median 21 vs. 9 Monate; p = 0,013; siehe Abb. 1) sowie beim rezidivfreien Überleben (Medianwert unter Sorafenib nicht erreicht vs. 23 Monate unter Placebo; p = 0,017). Beim Gesamtüberleben deutet sich ein numerischer Vorteil für Sorafenib an, ist aber noch nicht signifikant (nach drei Jahren 63% vs. 56%; p = 0,382). Die Kombination war gut verträglich, wenngleich unter Sorafenib häufiger Fieber, Blutungen und Hand-Fuß-Syndrome auftraten.
Damit wurde zum ersten Mal in einem randomisierten Setting bei der AML eine Verbesserung der Ansprechdauer mit einem Tyrosinkinaseinhibitor nachgewiesen. Andere spezifischere TKI sind für molekular definierte Subgruppen von Patienten in der Erprobung. Möglicherweise lassen sich auch in der SORAML-Population durch genetische Analyse Patienten identifizieren, die besser ansprechen als andere, sodass das Potenzial dieser Substanz damit noch optimiert werden kann.
Neuerungen auch bei klassischen Zytostatika
Dass auch bei den klassischen Zytostatika noch Fortschritte möglich sind, zeigte sich in der ebenfalls randomisierten und doppelblinden Phase-III-Studie VALOR, einer der bisher größten klinischen Studien zur rezidivierten oder refraktären AML [2]. Wie Farhad Ravandi, Houston, in der „Late-Breaking-Abstract“-Session erläuterte, erhielten die 711 Patienten Cytarabin und zusätzlich entweder Placebo oder den Topoisomerase-II-Hemmer Vosaroxin. Sehr ambitioniert wurde das Gesamtüberleben als primärer Endpunkt gewählt: Es war zwar in der Gesamtanalyse nicht signifikant unterschiedlich, aber in einer vorab geplanten Analyse, in der Stratifizierungsfaktoren wie Transplantationen berücksichtigt wurden, war Vosaroxin mit median 6,7 vs. 5,3 Monaten signifikant überlegen (p = 0,02). Das spiegelt sich auch in den Raten an Komplettremissionen (30,1% vs. 16,3%) und ebenso beim ereignisfreien und beim leukämiefreien Überleben wider. Die Toxizität im Verum-Arm war erwartungsgemäß höher, vor allem bedingt durch mehr Myelosuppression, was jedoch keinen Einfluss auf die Frühmortalität hatte, die nach 30 und 60 Tagen in beiden Armen ähnlich war. Eine Zulassung vorausgesetzt, bietet sich Vosaroxin/Cytarabin daher als neue Salvage-Option für diese Patienten an, für die wir bisher nur mehr wenige Möglichkeiten hatten.
Ebenfalls am M. D. Anderson Cancer Center wird Vosaroxin bei älteren Patienten (> 60 Jahre) mit neu diagnostizierter AML oder myelodysplastischem Syndrom mit hohem Risiko erprobt (d. h. mit mindestens 10% Blasten im Knochenmark). In einer Phase-I/II-Studie wurde der Topoisomeraseinhibitor mit dem demethylierenden Medikament Decitabin kombiniert [3]. Bei bisher 35 Patienten mit einem medianen Alter von 71 Jahren erwies sich diese Kombination als sehr wirksam, so Daver Naval, Houston – mit einer Gesamtansprechrate von 77%, davon 50% Komplettremissionen. Bei den 60–70-Jährigen war die Remissionsrate mit 88% deutlich höher als bei den älteren Patienten (71%). Besonders gut sprachen Patienten mit IDH2-Mutationen an (sieben von sieben Patienten).
MRD-Bestimmung bei NPM-mutierter AML
Die minimale Resterkrankung (MRD) entwickelt sich auch bei den akuten Leukämien zu einem immer wichtigeren Prognoseinstrument, das auch Einfluss auf Therapieentscheidungen nehmen kann. In einer britischen Studie wurden beispielsweise sequenzielle Blut- und Knochenmarksproben von Patienten mit AML und NPM1-Mutationen per Polymerasekettenreaktion (PCR) mit einer Sensitivität von 10-5 untersucht [4]. Patienten in morphologischer Komplettremission hatten eine deutlich schlechtere Prognose, wenn nach dem zweiten Chemotherapie-Zyklus noch MRD nachweisbar war, so Adam Ivey, London: Das Rezidivrisiko nach drei Jahren lag dann bei 77% gegenüber nur 28% bei MRD-negativen Patienten (p < 0,0001), und das wirkte sich direkt auf die 3-Jahres-Überlebensraten aus (25% vs. 77%; p < 0,0001).
Auch in einer multivariaten Analyse blieb der MRD-Status ein signifikanter Risikofaktor, während weitere ungünstige Prognosefaktoren wie FLT-ITD, DNMT3A und die konventionelle Risikoklassifikation ihren signifikanten Einfluss verloren. Die Ergebnisse der britischen Kollegen bedeuten eine dringende Empfehlung, die NPM1-PCR in die Routineversorgung aufzunehmen, weil sich daraus unmittelbar therapeutische Konsequenzen wie die Empfehlung zu einer Stammzelltransplantation ableiten lassen, bei der bisher neben Alter und Komorbiditäten vor allem Faktoren wie zytogenetische und molekulare Risikogruppen berücksichtigt werden.
Auch Patienten mit sekundärer AML profitieren von Transplantation
Ob Patienten mit sekundärer AML (verursacht durch zytotoxische Therapien oder als Folge anderer hämatologischer Erkrankungen wie eines myelodysplastischen Syndroms) von einer allogenen Stammzelltransplantation in der ersten Komplettremission profitieren, war bisher nicht klar. In einer Registerstudie fanden schwedische Kollegen unter 5.881 Patienten, deren AML zwischen 1997 und 2013 behandelt worden war, 550 Patienten (9%) mit therapiebedingter und 1.098 (19%) mit AML nach anderen hämatologischen Erkrankungen [5]. Sowohl Patienten mit De-novo- wie auch solche mit sekundärer AML profitierten von einer Stammzelltransplantation stärker als von einer konventionellen Postremissions-Therapie, wie Christer Nilsson, Stockholm, zeigen konnte: De-novo-Patienten waren sieben Jahre nach Transplantation zu 60% am Leben, nach konventioneller Behandlung nur noch zu 44%, bei denen mit sekundärer Leukämie waren es 46% vs. 21%; das entspricht im letzteren Fall einer Hazard Ratio von 0,45, d. h. mehr als einer Halbierung des Mortalitätsrisikos. Diese Ergebnisse sind unmittelbar relevant für die Praxis: Immerhin weist von unseren AML-Patienten mehr als ein Viertel eine sekundäre Erkrankung auf.
Akute Promyelozyten-Leukämie (APL)
Die akute Promyelozyten-Leukämie (APL) ist durch die Einführung von Retinoiden und Arsentrioxid von einer hochgradig letalen zu einer in den meisten Fällen heilbaren Erkrankung geworden. Arsentrioxid (ATO) ist bisher nur im Rezidiv zugelassen, aber eine deutsch-italienische Phase-III-Studie zeigte, dass die Chemotherapie-freie Kombination aus all-trans-Retinsäure (ATRA) und ATO in der Erstlinientherapie von Niedrigrisiko-Patienten (d. h. < 10.000 Leukozyten/ml) mindestens so wirksam ist wie ATRA mit einem Anthrazyklin [6]. Uwe Platzbecker, Dresden, konnte in San Francisco die endgültige Analyse mit sämtlichen 254 Patienten nach median drei Jahren präsentieren [7], und darin wurde die Überlegenheit des Chemotherapie-freien Protokolls erneut bestätigt, mit signifikanten Unterschieden nach zwei Jahren bei ereignisfreiem Überleben (98% vs. 84,9%; p = 0,0002), kumulativer Rezidivrate (1,1% vs. 9,4%; p = 0,005) und Gesamtüberleben (99,1% vs. 94,4%; p = 0,01). ATRA-ATO ist damit eindeutig ein neuer Standard in der First-line-Therapie von Niedrigrisiko-Patienten mit APL.
Bei Patienten mit hohem Risiko ist die Lage bisher weniger klar. Die austral-asiatische Leukämie- und Lymphom-Gruppe (ALLG) hat in einer Phase-II-Studie ein Kollektiv von 124 Patienten aus allen Risikokategorien mit einem Protokoll behandelt, in dem zu ATRA in Induktion und Konsolidierung das Arsen hinzugegeben und dafür Idarubicin auf die Induktion alleine beschränkt wurde. Die 5-Jahres-Ergebnisse, die Harry Iland, Camperdown, vorstellte [8], zeigen Raten für das ereignisfreie Überleben von 90%, für das krankheitsfreie Überleben von 95%, für das Gesamtüberleben von 94% und für das kumulative Rezidivrisiko von 5%. Sie sind für Niedrigrisiko-Patienten günstiger, aber auch für die Hochrisiko-Subgruppe, die ungefähr ein Fünftel aller Patienten ausmachte, waren die Werte mit 83%, 95%, 87% und 5% ausgezeichnet. Im Vergleich mit den Ergebnissen der vorangegangenen austral-asiatischen Studie, in der nur ATRA und Idarubicin gegeben worden waren, erscheinen sämtliche Überlebensdaten signifikant überlegen.
Akute lymphatische Leukämie (ALL)
Die pädiatrischen Onkologen haben bei der kindlichen akuten lymphoblastischen Leukämie (ALL) große Erfolge erzielt und können mittlerweile die meisten Kinder heilen. Weniger gut sieht es bei den T-Zell-Leukämien dieses Typs aus, die häufig rezidivieren und dann eine schlechte Prognose haben, insbesondere wenn sie einen bestimmten Immunphänotyp, den „early thymic precursor“-Typ (ETP) aufweisen. Um hier einen individualisierten Therapieansatz zu überprüfen, schloss die US-amerikanische Children´s Oncology Group (COG) 1.144 Kinder mit T-ALL in eine Studie ein, von denen 11,3% den ETP-Immunphänotyp und weitere 17% einen ähnlichen Phänotyp („near-ETP“) zeigten [9]. Nach einer konventionellen Induktionstherapie wurden die Patienten anhand ihrer an Tag 8 im peripheren Blut und an Tag 29 im Knochenmark gemessenen minimalen Resterkrankung (MRD) in drei Strata aufgeteilt (Niedrigrisiko: MRD < 0,1%, intermediär: MRD 0,1% – < 1%, Hochrisiko: MRD > 1%). Intermediär- und Hochrisiko-Patienten erhielten eine zusätzliche kraniale Bestrahlung und wurden randomisiert, zusätzlich zur Post-Induktionstherapie Nelarabin oder keine zusätzliche Behandlung zu erhalten.
Zunächst zeigte sich, dass Kinder mit ETP- und „near-ETP“-Phänotyp nach der Induktion häufiger eine minimale Resterkrankung aufwiesen und meist in die höheren Risikogruppen fielen. Das wurde aber offenbar durch die personalisierte Fortführung der Therapie kompensiert, denn die Gesamtüberlebensraten nach fünf Jahren waren nicht unterschiedlich: ETP 93%, „near-ETP“ 91,6%, kein ETP 92%. Damit eröffnet sich zum ersten Mal eine Möglichkeit, die Prognose dieser kindlichen Hochrisiko-Patienten deutlich zu verbessern.
Pädiatrische Protokolle auch für ältere Patienten?
ALL-Patienten, die als „Heranwachsende“ oder „junge Erwachsene“ bezeichnet werden (im Alter zwischen 16 und etwa 39 Jahren) erhalten regelmäßig Therapieprotokolle, wie sie bei erwachsenen Patienten gegeben werden und die weniger intensiv, aber auch weniger wirksam sind als die typischen pädiatrischen Regime. In einer Phase-II-Studie mehrerer US-amerikanischer Studiengruppen wurden deshalb 296 Patienten dieser Altersgruppe mit einem pädiatrischen Standardprotokoll mit vier Zyklen intensiver Chemotherapie behandelt [10]. Das führte in der einarmigen Studie nach zwei Jahren zu einer ereignisfreien Überlebensrate von 66% und einer Gesamtüberlebensrate von 79%, so Wendy Stock, Chicago. Im Vergleich mit einer früheren Studie der Cancer and Leukemia Group B (CALGB), in der das ereignisfreie Überlebens bei 34% gelegen hatte, ist das fast eine Verdoppelung. In einer multivariaten Analyse erwiesen sich eine Leukozyten-Zahl von mehr als 30.000/µl und das Vorliegen von BCR-ABL als negative prognostische Faktoren, während MRD-Negativität nach der Induktionstherapie mit ausgezeichneten Resultaten assoziiert war.
Ph+-ALL mit Zweitgenerations-TKI behandeln
ALL hat im Vergleich zu anderen Formen dieser Erkrankung eine schlechte Prognose. Die Tyrosinkinaseinhibitoren, die primär zur Therapie der CML entwickelt wurden, bieten sich auch hier an, und in zwei Studien, die in San Francisco vorgestellt wurden, kamen Zweitgenerations-Substanzen zum Einsatz:
In einem Protokoll der italienischen GIMEMA-Gruppe wurde Dasatinib gegeben, das zu einem schnellen Abfall der Blasten führte [11]. 18,6% der 60 auswertbaren Patienten erzielten eine komplette molekulare Remission (d. h. keine nachweisbaren BCR-ABL-Transkripte) und erhielten für sechs weitere Monate Dasatinib, so Sabina Chiaretti, Rom, die übrigen bekamen, wenn sie zumindest eine komplette hämatologische Remission erreicht hatten, eine allogene Stammzelltransplantation bzw., wenn kein Spender zur Verfügung stand, einen Konsolidierungszyklus mit Clofarabin und Cyclophosphamid.
In einem von Deutschland aus organisierten europäischen Projekt [12] wurde der Zweitgenerationsinhibitor Nilotinib gegeben, kombiniert in der Induktion mit Vincristin und Dexamethason, in der Konsolidierung mit Methotrexat, Asparaginase und Cytarabin und in der Erhaltungstherapie mit 6-Mercaptopurin und Methotrexat bzw. Dexamethason und Vincristin. Eine 2-Jahres-Überlebensrate von 73% ohne Stammzelltransplantation ist bei den über 55-jährigen Patienten vielversprechend, so Oliver Ottmann, Frankfurt. Von 36 auswertbaren Patienten hatten alle bis auf einen eine komplette Remission erzielt, die Rate an MRD-Negativität (kein nachweisbares BCR-ABL) stieg von 11,4% nach der Induktion auf 26,3% während der Konsolidierung an; 80% der Patienten hatten zu diesem Zeitpunkt eine gute molekulare Remission. Die Toxizität war akzeptabel, und das macht die Behandlung mit Tyrosinkinaseinhibitoren zu einer attraktiven Option gerade für ältere Patienten mit Ph+-ALL.
Immuntherapien gegen akute Leukämien
Die genetische Heterogenität der akuten Leukämien ruft einerseits nach individualisierten Therapieansätzen, die spezifisch an den jeweiligen pathogenetischen Mechanismen angreifen, andererseits zeigt sich bei den soliden Tumoren zunehmend, dass Immuntherapien Krebszellen ohne Rücksicht auf deren Genetik angreifen und vernichten können. Dieser Ansatz ist nun auch in der Hämatologie angekommen, wie eine Reihe von Präsentationen auch zu akuten Leukämien in San Francisco zeigt.
Patienten mit ALL, die nach der Induktionstherapie zwar eine hämatologische Komplettremission, aber noch messbare minimale Resterkrankung (MRD) aufweisen, haben ein erhöhtes Rezidivrisiko und eine schlechtere Prognose. Bei diesen Patienten geht es in erster Linie darum, ein hämatologisches Rezidiv zu vermeiden, die MRD-Last zu senken und die Zeit bis zu einer allogenen Stammzelltransplantation zu überbrücken. Der bispezifische Antikörper Blinatumomab, der das CD19-Antigen auf leukämischen Blasten und das CD3-Antigen auf T-Zellen erkennt und die T-Zellen dadurch in engen Kontakt mit den Blasten bringt, hat in einer kleinen Phase-II-Studie mit 21 solchen Patienten bei 18 von ihnen zu einer MRD-Eliminierung geführt. Die BLAST-Studie [13], die Nikola Gökbuget, Frankfurt, vorstellte, sollte diese Ergebnisse an einem größeren Kollektiv von 116 ALL-Patienten, die nach mindestens drei intensiven Chemotherapien in einer MRD-positiven kompletten Remission waren, bestätigen. Die Rate an kompletten MRD-Eradikationen lag bei 78% und war von der Zahl der Vorbehandlungen abhängig: Wurden die Patienten in der ersten Komplettremission behandelt, wurden 82% MRD-negativ, in der zweiten Komplettremission waren es 71% und in der dritten nur mehr 50%. 98% der Patienten mit einen kompletten MRD-Remission erreichten diese bereits im ersten Blinatumomab-Zyklus. Dieser bispezifische Antikörper hat mit Sicherheit ein sehr hohes Potenzial, die Prognose von ALL-Patienten mit minimaler Resterkrankung wesentlich zu verbessern.
CTL019: Komplette Remissionen bei Kindern mit refraktärer ALL
Ein besonders vielversprechender immuntherapeutischer Ansatz scheinen T-Lymphozyten mit chimärem Antigenrezeptor (CAR-T-Zellen) zu sein. Die ersten in klinischer Entwicklung befindlichen CAR-T-Zellen richten sich ebenfalls gegen das CD19-Antigen auf den Zellen maligner B-Zellen. Das Prinzip: Den Patienten werden für diese Therapie T-Lymphozyten entnommen, denen man einen gentechnisch konstruierten Rezeptor einpflanzt, der sich zum einen gegen das CD19-Antigen richtet und andererseits in der T-Zelle Signalwege anstößt, die ihre Vermehrung und ihr aggressives Vorgehen gegen die CD19-positiven Zellen stimulieren. Stephan Grupp, Philadelphia, behandelte bisher 39 pädiatrische Patienten mit refraktärer ALL mit seinen CTL019-Zellen [14]. Von ihnen konnten 36 damit eine komplette Remission erzielen (92%).
War die Behandlung anfangs als Überbrückung zur allogenen Stammzelltransplantation gedacht, so hat sich inzwischen gezeigt, dass die meisten Patienten auch ohne weitere Therapie in einer anhaltenden Remission bleiben: Die mediane Beobachtungszeit ist zwar mit sechs Monaten noch relativ kurz, aber nach dieser Frist sind 70% der Patienten ereignisfrei am Leben, d. h. sie haben kein Krankheitsrezidiv erlitten – und das in den allermeisten Fällen ohne zusätzliche Behandlungen wie eine Transplantation. Bei diesen „Langzeit“-Respondern können CTL019-Zellen noch bis zu 30 Monate lang im Blut nachgewiesen werden. Sie verursachen eine B-Zell-Aplasie, weshalb bei den Patienten Immunglobuline substituiert werden müssen.
Diese Ergebnisse machen deutlich, warum CAR-T-Zellen als einer der vielversprechendsten neuen Therapieansätze in der Hämatologie gelten. In San Francisco wurden eine Reihe weiterer positiver Resultate mit CD19-spezifischen Zellen vorgeführt, aber das dürfte erst der Anfang sein: Natürlich lässt sich das Prinzip auf jedes andere Oberflächenantigen anwenden, sofern dessen Expression sich weitgehend auf die malignen Zellen beschränkt.
Literatur
1. Röllig C et al. ASH 2014, Abstract #6.
2. Ravandi F et al. ASH 2014, Abstract #LBA-6.
3. Naval D et al. ASH 2014, Abstract #385.
4. Ivey A et al. ASH 2014, Abstract #70.
5. Nilsson C et al. ASH 2014, Abstract #281.
6. Lo-Coco F et al. N Engl J Med 2013; 369: 11-21.
7. Platzbecker U et al. ASH 2014, Abstract #12.
8. Iland HJ et al. ASH 2014, Abstract #375.
9. Wood BL et al. ASH 2014, Abstract #1.
10. Stock W et al. ASH 2014, Abstract #796.
11. Chiaretti S et al. ASH 2014, Abstract #797.
12. Ottmann OG et al. ASH 2014, Abstract #798.
13. Goekbuget N et al. ASH 2014, Abstract #379.
14. Grupp SA et al. ASH 2014, Abstract #380.

Prof. Dr. med. Karl-Anton Kreuzer
Klinik I für Innere Medizin
Universitätsklinikum Köln
Kerpener Straße 62
50937 Köln
+49 221 47897626
+49 221 47897627
karl-anton.kreuzer[at]uni-koeln[dot]de