Histopathologie des adulten Weichteilsarkoms – Stellenwert der neuen WHO-Klassifikation
Gellért Bakos, Bence Sipos
Die 2013 publizierte aktuelle WHO-Klassifikation der Weichteil- und Knochentumoren [1] zeigt signifikante Erweiterungen im Vergleich zur Ausgabe von 2002. Es wurden Entitäten wie der gastrointestinale Stromatumor, die peripheren Nervenscheidentumoren und kutane Weichteiltumoren mit aufgenommen, die früher unter den Tumoren des Gastrointestinaltraktes, des Nervenssystems und der Haut in differenten Bänden der WHO-Klassifikation dargelegt wurden. Die Fortschritte der Molekulargenetik haben in vielen Bereichen eine Verfeinerung der diagnostischen Möglichkeiten des Histopathologen durch Etablierung immer spezifischerer molekularpathologischer und immunhistochemischer Marker mit sich gebracht.
Vor allen Dingen ist die immer besser reproduzierbare diagnostische Entitäts-Zuordnung zu erwähnen, die z.T. auch an sehr eingeschränkt repräsentativen bioptischen Proben möglich ist. Der klinisch tätige Kollege muss aber damit rechnen, dass eine eindeutige Entitäts-spezifische Zuordnung am bioptischen Material, wenn morphologisch richtungweisende Merkmale nicht erfasst sind, auch mit Zusatzmethoden nicht immer gelingt.
Eine spezielle Problematik verbirgt sich insbesondere hinter dem Grading der Sarkome, das meist nach dem
FNCLCC-Schema auf der Beurteilung des ganzen Tumors, v. a. der Nekroseausdehnung und der mitotischen Aktivität der mitosereichsten Areale basiert und auch eine Entitäts-Zuordnung voraussetzt. Eine Anwendung dieses Grading-Schemas auf kleine Biopsate ist nur bedingt möglich. Wegen der therapeutischen Relevanz wird trotz alledem klinischerseits eine Graduierung auch am Biopsiematerial gefordert und vorgenommen, wobei FNCLCC Grad 1 als „low-grade“, die Grade 2 und 3 als „high-grade“ zusammengefasst werden.
Im Weiteren werden wir häufigere und weniger häufige, aber diagnostisch relevante bzw. einzelne in die neue WHO Klassifikation erstmalig aufgenommene Entitäten und Tumorgruppen, einschließlich deren immunhistochemischer und molekularpathologischer Charakteristika, exemplarisch vorstellen. Dabei werden wir auf die in den letzten Jahren eingeführten neuen diagnostischen Marker besonders eingehen.
Adipozytäre Tumoren
Atypischer lipomatöser Tumor
Die Klassifikation der Tumoren des Fettgewebes wurde in der neuen WHO-Klassifikation wenig verändert. Die Kategorie des atypischen lipomatösen Tumors/hochdifferenzierten Liposarkoms wurde unter dem Terminus atypischer lipomatöser Tumor beibehalten mit einem Kommentar, dass der Tumor in den meisten adäquat resezierten Fällen keinen malignen Verlauf zeigt. In den Lokalisationen (insbesondere Retroperitoneum, Mediastinum, Samenstrang), wo eine komplette Entfernung nicht möglich ist, sind Rezidive oftmals vorprogrammiert und es kann auch ohne Dedifferenzierung oder Metastasen zu tumorbedingtem Ableben des Patienten kommen, was die Verwendung des Begriffes „hochdifferenziertes Liposarkom“ weiterhin rechtfertigt. Morphologisch, immunhistochemisch und molekularpathologisch sind die Entitäten „atypischer lipomatöser Tumor“ und „hochdifferenziertes Liposarkom“ nicht zu trennen.
Die morphologische Definition basiert auf dem Nachweis sogenannter atypischer Stromazellen, also oftmals spindeliger Zellen mit Hyperchromasie und Kern-Irregularitäten. Da diese manchmal nur spärlich eingestreut sind, ist die Beurteilung des Resektionsrands, wenn keine eindeutige Kapsel oder kein breiter Saum von normalem Gewebe vorhanden ist, oftmals schwierig, zumal die die Tumormasse bildenden Adipozyten nicht von normalen Fettgewebszellen zu unterscheiden sind. Entgegen der weitläufigen Meinung ist das Vorhandensein von „Lipoblasten“ weder erforderlich für die Diagnose noch stellen diese allein ein Malignitätskriterium bei sonst Lipom-ähnlicher Tumormorphologie dar. Charakteristisch ist das Vorhandensein einer MDM2- bzw. CDK4- Amplifikation, welche insbesondere bei morphologisch fraglichen Fällen durch Fluoreszenz-In-situ-Hybridisierung (FISH) nachgewiesen wird.
Inzwischen stehen auch gute immunhistochemische Surrogatmarker zur Verfügung (p16, CDK4 und mdm2), welche alternativ oder ergänzend herangezogen werden können. Die Auswertung der Immunhistochemie setzt eine gewisse Erfahrung und gut eingestellte Antikörper voraus, zumal auch infiltrierende Makrophagen eine positive Reaktion zeigen können.
Eine molekularpathologische Untersuchung wird von leitenden Autoritäten der Sarkom-Pathologie auch empfohlen, wenn eindeutige Atypien fehlen, aber entweder ein Rezidivtumor vorliegt oder die Tumorgröße (über
15 cm) oder die Lage (intraabdominal/retroperitoneal) für ein Lipom ungewöhnlich ist [2].
Dedifferenziertes Liposarkom
Die Definition der dedifferenzierten Liposarkome (DDLPS) befindet sich auch im Wandel. Morphologisch wurde diese Entität ursprünglich definiert als atypischer lipomatöser Tumor/hochdifferenziertes Liposarkom mit Übergang in ein nicht lipogenes „high-grade“-Sarkom. Inzwischen sind mehrere Tumoren publiziert, wo die dedifferenzierte Komponente durchaus lipogene Eigenschaften, z. B. in Form eines pleomorphen Liposarkoms aufwies [3]. Weiterhin ist heute die Möglichkeit
einer Dedifferenzierung in eine „low-grade“-Komponente allgemein akzeptiert. Damit wurde in der neuen WHO-Klassifikation der gemischte Typ des Liposarkoms gestrichen. Diese Tumoren sind wohl seltene Varianten eines dedifferenzierten Liposarkoms.
Das typische Vorliegen einer MDM2-Amplifikation in DDLPS bietet auch die Möglichkeit, den Tumoren, bei denen der dedifferenzierte Tumoranteil den hochdifferenzierten überwuchert, oder in einer kleinen Biopsie nicht repräsentativ erfasst wird, durch Immunhistochemie oder FISH-Analyse auf die Schliche zu kommen. Dass das durchaus klinische Relevanz haben kann, zeigte kürzlich eine Untersuchung, in der der klinische Verlauf „undifferenzierter pleomorpher Sarkome mit MDM2-Amplifikation“ mit dem von dedifferenzierten Liposarkomen vergleichbar und deutlich besser war als der von undifferenzierten pleomorphen Sarkomen ohne MDM2-Amplifikation (2-Jahres-Gesamtüberleben von 93,3% und 90,7% versus 73,9%). Dies wäre ein Argument, undifferenziert erscheinende Sarkome mit MDM2-Amplifikation als DDLPS einzuordnen, was sich allerdings dann trotz der Morphologie auch auf das Grading des Tumors (Grad 2 statt 3) auswirken würde. Bemerkenswert ist, dass die Prognose in erster Linie von der Lokalisation des Tumors abhängig zu sein scheint und dass weder die Ausdehnung noch der Grad der dedifferenzierten Komponente wesentlichen Einfluss hat. Dies bedeutet auch, dass viele Experten den DDLPS biologisch für ein „Grad 2“-Sarkom halten, ohne Rücksicht auf den Grad („low“- vs. „high-grade“) des dedifferenzierten Tumoranteils.
Myxoides Liposarkom
Myxoide Liposarkome (MLS) sind überwiegend Tumoren des tiefen Weichgewebes der Extremitäten und entstehen am häufigsten in der Oberschenkelmuskulatur. Das Metastasierungsmuster ist oft ungewöhnlich, wobei Weichgewebe, Knochen und seröse Häute befallen sein können, auch ohne dass Lungenmetastasen vorliegen [4].
Charakteristisch sind myxoide Bezirke mit geringer Zellularität und gebogenen, teils „hahnentrittartigen“ Kapillaren. Prognoserelevant sind und für das Grading verwendet werden sogenannte „rundzellige Areale“, wo die Tumorzellen dicht beieinander liegend rasenartige Bezirke bilden [5]. Der Grenzwert für das Ausmaß der rundzelligen Areale als Differenzierungsmerkmal zwischen „low“- und „high-grade“-Tumoren variiert in verschiedenen Studien zwischen 5% und 25% [4], sodass die Tumormasse bei „high-grade“-MLS überwiegend eine „low-grade“-Morphologie haben kann, was bei bioptischem Material die Wahrscheinlichkeit eines Sampling-Errors erhöht. Hervorzuheben ist, dass diese „rundzelligen Areale“ eine histologische Variante darstellen, die bei dem sonst üblichen FNCLCC-Grading nicht berücksichtigt wird. Der alte Begriff „Rundzell-Liposarkom“ wird in der neuen Klassifikation aufgehoben, und unter den Synonymen dem „high-grade“-MLS zugeordnet [5]. Insbesondere wenig differenzierte, rundzellreiche Varianten (oder kleine Biopsate mit gleichartigen, eingeschränkt repräsentativen Arealen) können diagnostische Schwierigkeiten bereiten. Bei einer klein-blau-rundzelligen Morphologie kann eine fokale S100-Positivität den ersten Hinweis geben. Die Abgrenzung gegenüber anderen klein-blau-rundzelligen Tumoren kann unter Umständen nur durch den Nachweis des charakteristischen DDIT3 (CHOP)-Rearrangements (zu ca. 95% Fusion mit dem FUS-Gen, zu ca. 5% mit dem EWSR1-Gen) erfolgen [4].
Pleomorphes Liposarkom (PLS)
Pleomorphe Liposarkome stellen die seltenste Liposarkom-Gruppe dar. Sie werden morphologisch durch den Nachweis oft atypischer Lipoblasten von undifferenzierten pleomorphen Sarkomen unterschieden, was die Wichtigkeit einer extensiven Probenentnahme zur Diagnosesicherung unterstreicht. Besonders in kleineren Proben ist die Kenntnis über mögliche epitheloide Areale in PLS wichtig, da diese ohne erfasste Lipoblasten leicht zu einer Diagnose eines „epitheloiden Sarkoms“ im weitesten Sinne führen können. Es handelt sich meist um „high-grade“-Sarkome der Extremitäten, deren Diagnose grundsätzlich auf der Morphologie beruht. Eine S100-Immunhistochemie kann die eventuell nur spärlichen Lipoblasten hervorheben. Molekulargenetische Marker (MDM2-Amplifikation) haben nur insofern Relevanz, als dadurch Imitatoren, zum Beispiel homolog lipogen dedifferenzierte Liposarkome, differenziert werden können [4].
Leiomyosarkome
Die neue WHO-Klassifikation hat keine wesentlichen Änderungen in der Einordnung der leiomyogenen Sarkome gebracht [6]. Als relativ spezifischer leiomyogener immunhistochemischer Marker hat sich in den letzten Jahren h-Caldesmon erwiesen. Eine Positivität gegenüber mindestens zwei immunhistochemischen Markern und/oder typische leiomyogene faszikuläre Areale werden für die Diagnose gefordert. Die Abgrenzung von hochdifferenzierten Leiomyosarkomen gegenüber gutartigen Leiomyomen kann bei eingeschränkt repräsentativer Probe problematisch sein.
Rhabdomyosarkome
Embryonales und alveoläres Rhabdomyosarkom
Rhabdomyosarkome (RMS) stellen die häufigsten Sarkome des Kindesalters dar und sind bei Erwachsenen selten. In der pädiatrischen Population sind insbesondere das embryonale RMS (ERMS) und das alveoläre RMS (ARMS) erwähnenswert [7]. Da besonders ARMS auch bei jungen Erwachsenen auftreten kann, wird auf diese Entitäten eingegangen.
Morphologisch zeigen klassische embryonale RMS unterschiedliche Reifestadien der Rhabdomyogenese, wobei das histologische Bild in Abhängigkeit vom vorherrschenden Zelltyp recht unterschiedlich sein kann. Treten pleomorphe Tumorzellen oft mit assoziierten Mitosen auf, spricht man von der anaplastischen Variante des ERMS. Sind die Proliferate assoziiert mit der Schleimhaut und entsteht dabei eine subepitheliale Kambiumschicht-artige Tumorzellverdichtung, oft mit polypoider, teils traubenartig vorwölbender Tumormasse, spricht man von einer botryoiden Variante. Typisch ist eine Expression der myogenen Marker, wobei neben Desmin insbesondere die für die skelettmuskuläre Differenzierung spezifischere Myogenin-Expression von Bedeutung ist.
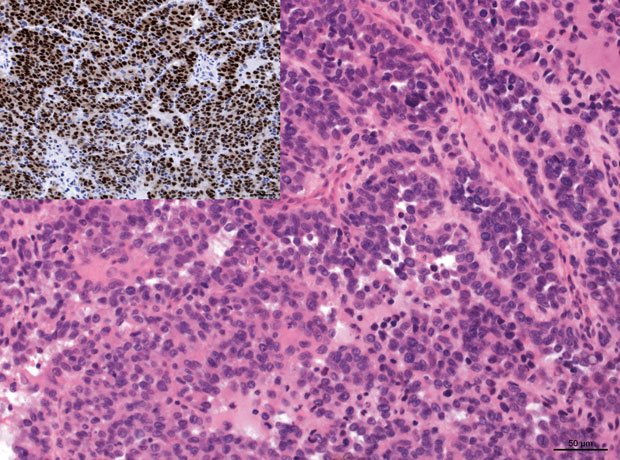
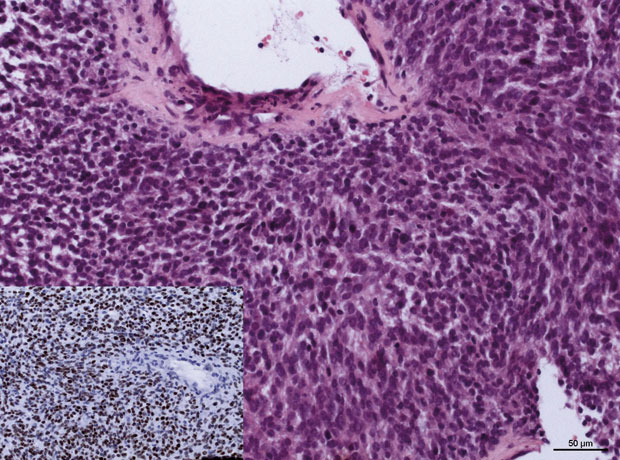
Das ARMS ist ein agressiver Tumor, der zunächst anhand der morphologischen Merkmale definiert und bezeichnet wurde. Es liegt eine monomorphe, relativ kleinzellige Tumorpopulation vor, die in klassischen Fällen durch Bindegewebs-Septen in Tumorzellgruppen unterteilt wird (Abb.1). Auch die klassischen Formen zeigen oftmals Bezirke ohne Septen-Bildung, d. h. ausgedehnte Tumorzellrasen in solider Anordnung. Fehlt die septale Untergliederung, d. h. die alveoläre Struktur komplett, spricht man von der soliden Variante des ARMS.
Obwohl feine zytologische Unterschiede oft bestehen, bedarf eine sichere Differenzierung dieser Variante von anderen klein-blau-rundzelligen Tumoren in der Praxis weiterer immunhistochemischer Analysen. Bei immunhistochemischen Hinweisen auf eine rhabdomyogene Differenzierung kann oft das Expressionsmuster des Myogenins weiterhelfen. Dieses ist in ERMS typischerweise nur fokal, in ARMS hingegen diffus und kräftig (Abb.1). Den molekularpathologischen Beweis eines ARMS bringt der Nachweis eines FOXO1-Rearrangements als Folge von Translokationen, die zu den Fusionen PAX3-FOXO1 bzw. PAX7-FOXO1 führen.
Leider ist die Klassifikation nicht immer eindeutig, zumal ca. 20–30% der ARMS PAX-Fusion-negativ sind. Die Untersuchungen von Davicioni et al. und Williamson et al. ergaben [8, 9], dass Fusions-negative ARMS weder auf molekularer Ebene noch im klinischen Verlauf von ERMS zu differenzieren sind. Auch wenn man bedenkt, dass seltene alternative Genfusionen durch die in der Routinediagnostik verwendeten FOXO1-Rearrangement-Kits nicht erfasst werden und gelegentliche ERMS/ARMS-Hybridtumoren existieren, besteht durchaus die Möglichkeit, dass ERMS (zyto-)morphologisch ARMS imitieren können. Da durch die Verwendung der molekularpathologischen Zusatzanalysen die morphologische Definition des ARMS ins Wanken geraten ist, gibt es Tendenzen, genetischen Klassifikation für die Prognoseabschätzung und Risikoeinstufung gegenüber der Histomorphologie den Vorzug zu geben.
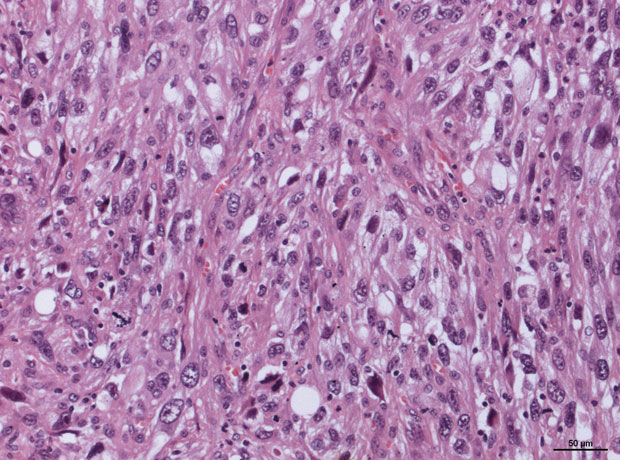
Pleomorphes Rhabdomyosarkom
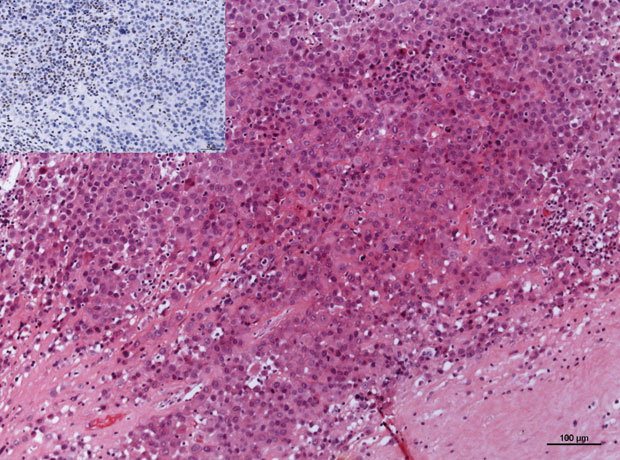
Pleomorphe Rhabdomyosarkome (Abb. 2) sind demgegenüber meist Tumoren des Erwachsenenalters und treten oft im Bereich der Extremitäten auf [10]. Sie werden morphologisch definiert als „high-grade“-Sarkome aus teils bizarren Tumorzellen mit rhabdomyogener Differenzierung ohne Komponenten eines ERMS oder ARMS. Neben den bizarr pleomorph-zelligen Tumoren, kommen auch spindelzellig-faszikuläre und rundzellig dominierte morphologische Varianten vor. Die rhabdomyogene Differenzierung bedeutet den morphologischen Nachweis von Rhabdomyoblasten, der in praxi immunhistochemisch durch myogene Marker,
z. B. Desmin (höhere Sensitivität, aber nicht spezifisch für quergestreifte Muskulatur) und relative Skelettmuskel-spezifische Marker wie Myogenin (myf4), MyoD1, evtl. Myoglobin sowie sogenanntes „fast myosin“, untermauert wird.
Für die Diagnosestellung wird als Mindestkriterium die Positivität von mindestens einem rhabdomyogenen Marker neben der Expression eines weiteren myogenen Markers gefordert. Allerdings besteht bis heute kein allgemeiner Konsens, wie viele und welche Marker eingesetzt werden sollen, um eine rhabdomyogene Differenzierung zu beweisen/auszuschließen. Pleomorphe Rhabdomyosarkome zeigen gegenüber anderen pleomorphen Sarkomen eine schlechtere Prognose, was die Relevanz der Zusatzuntersuchungen bei pleomophen „high-grade“-Sarkomen unterstreicht.
Seltene Rhabdomyosarkom-Varianten
stellen das sklerosierende RMS und das spindelzellige RMS dar, die im aktuellen WHO-Band [1] als „spindelzelliges/sklerosierendes RMS“ mit soweit unklarem nosologischem Status klassifiziert werden. Die Tumoren sind bei Jungen/Männern auffallend häufiger und treten bei Kindern insbesondere im para-testikulären Bereich auf, bei einem 5-Jahres-Überleben von 95%. Bei Erwachsenen liegt meist ein Tumor im Kopf-Halsbereich vor, die Prognose ist dabei deutlich schlechter.
Eine kürzlich beschriebene Rhabdomyosarkom-Variante stellt das epitheloide RMS [11] dar, das zunächst bei älteren Patienten, inzwischen aber auch bei Kindern beschrieben wurde. Wichtig ist dieser Phänotyp wegen der Karzinom-ähnlichen Morphologie, die mit zumindest fokaler Zytokeratin-Positivität gekoppelt sein und zu Fehldiagnosen führen kann. Obwohl die nosologische Zugehörigkeit noch unklar ist, gibt es in der pädiatrischen Population keine Hinweise auf ARMS-typische Fusionsprodukte [12].
Bemerkenswert ist weiterhin die rhabdomyogene Differenzierung als heterologes Element bei anderen Entitäten, z. B. dem dedifferenzierten Liposarkom, malignen peripheren Nervenscheidentumoren (maligner Triton-Tumor), dem malignen Müllerschen Mischtumor oder sekundär zum Beispiel auf dem Boden von Teratomen, was bei eingeschränkt repräsentativer Biopsie die abschließende Zuordnung eines myogen differenzierten Sarkoms weiter erschweren kann.
Fibroblastische/myofibroblastische Sarkome
Myxofibrosarkom
Myxofibrosarkome [13] gehören zu den häufigsten Sarkomen bei älteren Patienten (Gipfel zw. 50 und 80 Jahren). Überwiegend befinden sie sich an den Extremitäten. Dermale/subkutane Lokalisation kommt genauso häufig vor wie eine fasziale/subfasziale Lokalisation. Histomorphologisch zeigt sich ein oft multinodulärer Tumor, der insbesondere bei oberflächlicher Lokalisation ein deutlich infiltratives Wachstum aufweisen kann, das präoperativ oft unterschätzt wird. Morphologisch definierend ist eine lockere, myxoide Grundmatrix, in der die Tumorzellen unterschiedlich dicht eingestreut sein können. „Low-grade“-Myxofibrosarkome können, vor allem bei eingeschränkt repräsentativer Biopsie, harmlos aussehen. Mitosen sind selten, die zellulären Atypien teilweise diskret, Nekrosen fehlen, eine Abgrenzung gegenüber gutartigen myxoiden Tumoren kann durchaus Probleme bereiten. Eine für Myxome typische aktivierende Mutation des GNAS-Gens ist allerdings bei Myxofibrosarkomen nicht nachweisbar [14].
Die Diagnose der höhergradigen Myxofibrosarkome fällt meist nicht schwer, oft sind eine höhere Zellularität, deutlichere Atypien, Mitosen und Nekrosen kennzeichnend. Immunhistochemisch bestehen Hinweise auf myofibroblastäre Differenzierung. Glattmuskel-Aktin ist gelegentlich positiv, wohingegen Desmin und Marker einer weiteren Liniendifferenzierung nicht exprimiert werden.
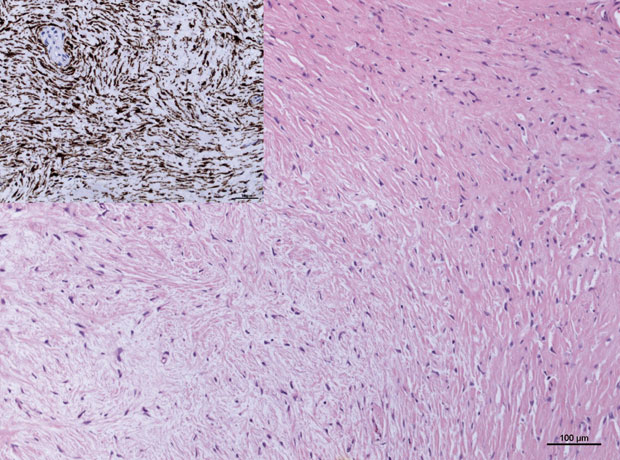
“Low-grade”-fibromyxoide Sarkome
sind relativ seltene Tumoren des meist jungen Erwachsenenalters, die mit dem Grad-1-Myxofibrosarkom verwechselt werden können [15]. Dieser Tumor ist meist subfaszial gelegen und befällt in den meisten Fällen die Extremitäten. Histopathologisch besteht eine sehr blande, als gutartig imponierende Zytomorphologie (Abb. 3). In der Übersichtsvergrößerung ist ein etwas nodulär imponierendes Nebeneinander von fibrösen und myxoiden Zonen charakteristisch mit geringer bis mäßiger Zellularität. Vage faszikuläre und wirbelige Zelllagerung, filigrane plexiforme Kapillaren mit teils perivaskulärer Sklerosierung kommen vor. Selten, aber typisch sind sogenannte kollagenöse Riesenrosetten, d. h. noduläre Bezirke aus zellarmem, homogenisiertem Kollagen. Die Diagnostik dieser Tumorentität wurde wesentlich erleichtert durch den Antikörper MUC4 (Abb. 3), der im entsprechenden morphologischen Kontext ein sensitiver und spezifischer Marker zu sein scheint [16].
Der molekularpathologische Nachweis (z. B. mittels FUS-FISH) einer konsistent zu beobachtenden Genfusion (FUS-CREB3L2, deutlich seltener FUS-CREB3L1 in insgesamt über 80–90% der Fälle) kann besonders bei atypischer Lokalisation die Diagnose erleichtern. Trotz der blanden Tumormorphologie zeigen diese Tumoren im Langzeitverlauf oft späte pleuropulmonale Metastasen mit tumorbedingtem Tod [15].
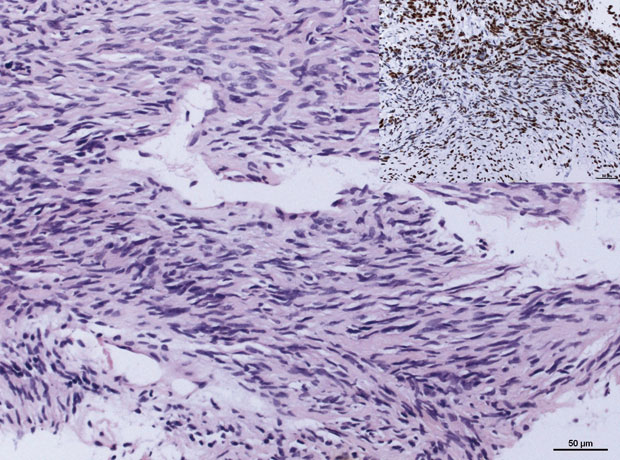
Extrapleuraler solitärer fibröser Tumor (SFT)
Der SFT, früher auch Hämangio-Perizytom genannt, ist ein Tumor des Erwachsenen, der inzwischen in nahezu allen Lokalisationen beschrieben worden ist und sowohl subkutan als auch im tiefen Weichgewebe auftreten kann. Eine paraneoplastische Hypoglykämie kann bei größeren Tumoren auftreten.
Morphologisch findet man einen meist scharf umschriebenen Tumor, der teils zellreiche, teils fibröse, sklerosierte und zellarme Areale aufweisen kann, mit typischen, aber an sich nicht diagnostischen ektatischen hirschgeweihartig verzweigenden Gefäßen (Hämangio-Perizytom-artiges Gefäßmuster, Abb. 4). Die Tumorzellen sind überwiegend spindelförmig und liegen meist ohne faszikuläre oder storiforme Architektur „strukturlos“ im Stroma. Perivaskuläre Hyalinisierung kann prominent sein. Mitosen sind meistens rar. Fettgewebs-reiche Varianten besonders im Retroperitoneum können in der Stanzbiopsie ein dedifferenziertes Liposarkom imitieren. Die Diagnose basierte in den letzten Jahren auf typischer Morphologie, verbunden mit der typischen diffusen Expression von CD34, oft mit Koexpression von Bcl2 und CD99. Fokale EMA-, SMA- und S100-Positivität kann vorkommen.
Das Verhalten der SFT ist meist gutartig, aber morphologisch nicht immer vorhersagbar. In ca. 10% der Fälle kann es zu teils späten Rezidiven oder Metastasen kommen. Maligne SFT (Abb. 5) haben teilweise typische morphologische Merkmale von Malignität, bei denen die Diagnose eines Sarkoms leichtfällt, die Entitätszuordnung bei oft deutlich reduzierter CD34-Expression aber eine subjektive Komponente auf Seite des Histopathologen beinhaltet. Der seit Kurzem auch in der diagnostischen Routine verwendete STAT6-Antikörper [17] als Surrogatmarker für eine NAB2-STAT6-Genfusion, der als spezifischer Marker des SFT angesehen wird, erscheint eine vielversprechende Möglichkeit, auch diese Tumoren reproduzierbar und an kleinen Proben richtig einzuordnen (Abb. 5).
Vaskuläre Tumoren
In die aktuelle WHO-Klassifikation der vaskulären Tumoren wurde die vor Kurzem beschriebene Entität des pseudomyogenen Hämangioendothelioms [18] aufgenommen. Es handelt sich um einen meist bei Teenagern und jungen Männern auftretenden Tumor, der häufig multizentrisch an den Extremitäten vorkommt und sowohl oberflächliches als auch tiefes Weichgewebe mit Knochen betreffen kann. Lokalrezidive sind häufig, Fernmetastasen selten (< 1% in den beschriebenen Fällen). Das lichtoptische Bild lässt den Pathologen in erster Linie an einen myogenen oder epithelialen Tumor denken. Das Pan-Cytokeratin (AE1/AE3) ist oft diffus positiv wie bei Karzinommetastasen, aber es findet sich eine Expression von Endothelmarkern, wie ERG, FLI-1, oft CD31. Ein weiterer Endothelmarker, CD34, der oft auch auf epitheloiden Sarkomen exprimiert wird, bleibt hier negativ, wie auch der myogene Marker Desmin. INI1 ist im Gegensatz zu epitheloiden Sarkomen nukleär erhalten. Eine charakteristische Translokation in diesen Tumoren t(7;19)(q22;q13) kann in Zukunft diagnostische Wertigkeit erlangen.
Epitheloide Hämangioendotheliome werden zusammen mit Angiosarkomen in der malignen Kategorie der vaskulären Tumoren geführt, obwohl epitheloide Hämangioendotheliome deutlich weniger aggressiv sind: In ca. 15% der Fälle kommt es zum tumorbedingten Tod. Ein höheres Risiko ist bei Tumoren über 3 cm mit mehr als drei Mitosen/50 HPF zu erwarten. Eine konsistent nachweisbare Translokation mit resultierender WWTR1-CAMTA1-Fusion scheint tumorspezifisch zu sein [19].
Die Diagnostik der Angiosarkome, die deutlich aggressiver sind, hat sich in der neuen WHO-Klassifikation nicht wesentlich geändert. Typisch sind morphologische Hinweise auf vaskuläre Differenzierung. Immunhistochemisch sind die Marker ERG, CD31, CD34, FLI1 richtungweisend. Eine fehlende HHV8-Assoziation hilft, Kaposi-Sarkome zu differenzieren. Eine partielle Cytokeratin-Expression kann unter Umständen zur Fehlinterpretation als Karzinom führen.
Maligner peripherer Nervenscheidentumor (MPNST)
Der MPNST ist ein Sarkom mit Nervenscheidendifferenzierung, das seinen Ursprung meist aus einem großen Nerven oder einem gutartigen Nervenscheidentumor, überwiegend einem Neurofibrom nimmt oder in einem Patienten mit Typ-1-Neurofibromatose entsteht. Außerhalb dieses Kontextes sind histologische und immunhistochemische Hinweise auf Schwannzell-Differenzierung gefordert. Meist liegt ein zellreiches spindelzelliges oder epitheloidzelliges Sarkom vor. Immunhistochemisch ist eine fokale (in epitheloiden Varianten auch diffuse) Positivität für S100 zu erwarten. Im Kontext der Weichteilsarkome scheint der Marker SOX10 spezifischer, aber wenig sensitiv zu sein [20]. In vielen typischen Fällen ist aber sowohl S100 als auch SOX10 negativ. Eine Abgrenzung gegenüber Melanom-Varianten kann unter Umständen problematisch sein. Rhabdomyosarkomatöse und angiosarkomatöse heterologe Differenzierung kann vorkommen.
Sarkome von unsicherer Histogenese
Synoviales Sarkom
Das synoviale Sarkom (SS, [21]) ist ein Tumor von unsicherer Histogenese, der unterschiedlich ausgeprägte epitheliale Differenzierung zeigen kann. Er kann vor allem bei spärlichem bioptischem Material viele Entitäten von Karzinomen bis hin zu klein-blau-rundzelligen Tumoren imitieren. Als Sarkom mit der höchsten Prävalenz bei Teenagern und jungen Erwachsenen kommt dieser Tumor nicht nur Gelenk-assoziiert, sondern in vielen anderen Lokalisationen einschließlich Retroperitoneum und retroperitonealer Organe vor. In der monophasischen Variante liegt ein meist zellreicher, spindelzelliger oder klein-blau-rundzelliger Tumor vor, wobei Hämangioperizytom-artige Gefäße und hyaline Regressionszonen nicht selten sind. Bei dem selteneren biphasischen Typ findet sich eine zusätzliche epitheliale Komponente, die des Öfteren drüsig differenziert ist und, sofern die Stromakomponente als desmoplastische Reaktion fehlinterpretiert werden sollte, auch falsch als Adenokarzinom eingestuft werden kann.
Eine Expression von Cytokeratinen und/oder EMA ist auch beim monophasischen Typ fokal oft vorhanden und lässt den Pathologen bei einem spindelzelligen sarkomatoiden Tumor auch an SS denken. Ergänzende Positivität für TLE1 ist typisch, aber leider nicht ganz spezifisch, zumal sie auch in MPNST und SFT beschrieben ist. Eine meist nur fokale S100-Positivität kann die Abgrenzung gegenüber neurogenen Tumoren erschweren. Eine CD99-Positivität kann bei entsprechender Morphologie eine Verwechslung mit dem Ewing-Sarkom begünstigen. In fraglichen und Cytokeratin-negativen Fällen hilft die molekularpathologische Analyse weiter mit der Demonstration eines Rearrangements (meist mittels FISH oder RT-PCR) als Folge der t(X;18)(p11;q11) Translokation mit resultierender SS18-SSX-Genfusion.
Die Prognose des Tumors korreliert mit Stadium und Tumorgrad. SS im Bereich der Extremitäten haben oft eine bessere Prognose.
Epitheloides Sarkom
Das epitheloide Sarkom (ES, [22]) ist ein typischer Fallstrick für den Histopathologen, mit zwei definierten Subtypen:
- Der klassische/distale Typ präsentiert sich meist bei Teenagern und jungen Erwachsenen an den distalen Extremitäten (oft im Bereich der Hand), als teils exulzerierende dermale, subkutane knotige Indurationen, die auch multipel auftreten können. Trotz der Bezeichnung können die Tumorzellen ausgesprochen Zytoplasma-arm und spindelig erscheinen oder an Aggregate bildende Makrophagen erinnern. Man kann in kleinen Proben den ersten histologischen Eindruck eines granulomatösen oder fibrosierenden Prozesses oder eines in Organisation begriffenen Hämatoms gewinnen.
- Der proximale Typ (Abb. 6) ist ein Tumor des Erwachsenenalters, der am häufigsten im tiefen Weichgewebe im Becken-Oberschenkelbereich, seltener auch in anderen Lokalisationen auftritt und aufgrund großer epitheloider, teils rhabdoider Tumorzellen zur Fehldiagnose eines Karzinoms oder RMS führen kann. Eine charakteristischerweise negative INI1-Reaktion und eine häufige Positivität gegenüber CD34 sind hilfreiche immunhistochemische Merkmale. Lokale Rezidive sind häufig und Metastasen in lokoregionären Lymphknoten und in der Lunge sind bei ca. 50% der Patienten zu erwarten.
Weitere Änderungen, neue Entitäten
Extraskelettale Ewing-Sarkome sind im Erwachsenenalter seltene klein-blau-rundzellige Sarkome mit typischer, aber nicht spezifischer membranöser CD99-Expression. FLI1 und ERG können positiv sein, CD56 und Myogenin sind negativ. In den meisten Fällen besteht eine EWSR1-Translokation, die meist in einer EWSR1-FLI1-Fusion (ca.85% der Fälle), seltener in einer Fusion mit anderen Genen der ETS-Genfamilie resultiert [23].
Das früher als Synonym verwendete PNET (peripherer/primitiver neuroektodermaler Tumor) wurde in die aktuelle Nomenklatur nicht übernommen, um Verwechslungen mit ähnlich bezeichneten Tumoren ohne EWSR1-Translokation im Zentralnervensystem und dem weiblichen Genitale zu vermeiden [6].
Eine wichtige, da ohne klinische Information schwer zu diagnostizierende, neu aufgenommene Entität ist der phosphaturische mesenchymale Tumor [6, 24], der typischerweise mit Osteomalazie einhergeht und sich nur selten maligne verhält. Eine Erhöhung von Fibroblast Growth Factor Typ 23 (FGF23) im Serum bei Osteomalazie kann die Diagnostik in die richtige Richtung lenken. Die Morphologie ist die eines meist aus blanden ovalen bis spindeligen Zellen bestehenden Tumors mit teils ungewöhnlicher fokal verkalkter, basophiler Matrix. Hämangioperizytom-artiges Gefäßmuster, Adipozyten und osteoklastäre Riesenzellen können in größerer Anzahl vorhanden sein und die histologische Differenzialdiagnostik bei Fehlen eines spezifischen Immunphänotyps deutlich erschweren.
Undifferenzierte/Unklassifizierte Sarkome
Als neue Kategorie der aktuellen WHO-Klassifikation umfasst diese Gruppe eine Reihe verschiedener Tumoren, die mit den heutigen Methoden nicht weiter klassifizierbar sind [1, 6]. Bestrahlungsassoziierte Sarkome sind nicht selten vom unklassifizierten Typ. Morphologisch sind vier Subgruppen auseinanderzuhalten.
1. Undifferenzierte pleomorphe Sarkome entsprechen im Wesentlichen den früheren klassischen pleomorphen malignen fibrösen Histiozytomen (MFH). Der Ausschluss einer MDM2/CDK4-Amplifikation, die bei gleicher Morphologie für ein dedifferenziertes Liposarkom sprechen würde, wird gefordert.
2. Undifferenzierte spindelzellige Sarkome.
3. Undifferenzierte rundzellige Sarkome, bei denen eine Verwandtschaft mit Ewing-Sarkomen nicht sicher ausgeschlossen ist, aber die klassischen Translokationen fehlen [25].
4. Epitheloidzellige Sarkome, die eine wenig charakterisierte Gruppe darstellen.
Literatur
1. Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds). WHO Classification of Tumours of Soft Tissue and Bone, 4th Edition. Lyon: IARC Press, 2013.
2. Zhang H et al. Molecular testing for lipomatous tumors: Critical analysis and test recommendations based on the analysis of 405 extremity-based tumors. Am J Surg Pathol 2010; 34: 1304-11.
3. Mariño-Enríquez A et al. Dedifferentiated liposarcoma with “homologous” lipoblastic (pleomorphic liposarcoma-like) differentiation: Clinicopathologic and molecular analysis of a series suggesting revised diagnostic criteria. Am J Surg Pathol 2010; 34: 1122-31.
4. Dei Tos A P. Liposarcomas: Diagnostic pitfalls and new insights. Histopathology 2014; 64: 38–52.
5. Antonescu CR, Ladanyi M. Myxoid liposarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds). WHO Classification of Tumours of Soft Tissue and Bone, 4th Edition. Lyon: IARC Press 2013: 39-41.
6. Fletcher CDM. The evolving classification of soft tissue tumours – an update based on the new 2013 WHO classification. Histopathology 2014; 64: 2–11.
7. Parham DM, Barr FG. Embryonal rhabdomyosarcoma. Alveolar rhabdomyosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds). WHO Classification of Tumours of Soft Tissue and Bone, 4th Edition. Lyon: IARC Press 2013: 127-32.
8. Davicioni E et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res 2006; 66: 6936-46.
9. Williamson D et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol 2010; 28: 2151-8.
10. Furlong MA et al. Pleomorphic rhabdomyosarcoma in adults: A clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol 2001; 14: 595-603.
11. Jo VY et al. Epithelioid rhabdomyosarcoma: Clinicopathologic analysis of 16 cases of a morphologically distinct variant of rhabdomyosarcoma. Am J Surg Pathol 2011; 35: 1523-30.
12. Zin A et al. Epithelioid rhabdomyosarcoma: A clinicopathologic and molecular study. Am J Surg Pathol 2014; 38: 273-8.
13. Mentzel T et al. Myxofibrosarcoma. Clinicopathologic analysis of 75 cases with emphasis on the low-grade variant. Am J Surg Pathol 1996; 20: 391-405.
14. Willems SM et al. Cellular/intramuscular myxoma and grade I myxofibrosarcoma are characterized by distinct genetic alterations and specific composition of their extracellular matrix. J Cell Mol Med 2009; 13: 1291-301.
15. Evans HL. Low-grade fibromyxoid sarcoma: A clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 2011; 35: 1450-62.
16. Doyle LA et al. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am J Surg Pathol 2011; 35: 733-41.
17. Doyle LA et al. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod Pathol 2014; 27: 390-5.
18. Hornick JL et al. Pseudomyogenic hemangioendothelioma: A distinctive, often multicentric tumor with indolent behavior. Am J Surg Pathol 2011; 35: 190-201.
19. Errani C et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 2011; 50: 644-53.
20. Karamchandani JR et al. Sox10 and S100 in the diagnosis of soft-tissue neoplasms. Appl Immunohistochem Mol Morphol 2012; 20: 445-50.
21. Suurmeijer AJH et al. Synovial sarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds). WHO Classification of Tumours of Soft Tissue and Bone, 4th Edition. Lyon: IARC Press 2013: 213-15.
22. Oda Y et al. Epitheloid sarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds). WHO Classification of Tumours of Soft Tissue and Bone, 4th Edition. Lyon: IARC Press 2013: 216-18.
23. Fisher C. The diversity of soft tissue tumours with EWSR1 gene rearrangements: A review. Histopathology 2014; 64: 134–50.
24. Folpe AL. Phosphaturic mesenchymal tumour. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds). WHO Classification of Tumours of Soft Tissue and Bone, 4th Edition. Lyon: IARC Press 2013: 211-12.
25. Antonescu C. Round cell sarcomas beyond Ewing: Emerging entities. Histopathology 2014; 64: 26–37.

Dr. Gellért Bakos
Institut für Pathologie und Neuropathologie, Abteilung Allgemeine Pathologie, UKT
Eberhard-Karls-Universität Tübingen
Liebermeisterstr. 8, 72076 Tübingen