Die Therapie maligner Non-Hodgkin-Lymphome (NHL) hat in den vergangenen 15 Jahren Fortschritte gemacht, vor allem durch die Einführung von CD20-Antikörpern. Diese Entwicklung setzt sich fort: Soeben ist der Typ-II-Antikörper Obinutuzumab mit veränderter Glykosylierung zur Behandlung älterer, gesundheitlich kompromittierter Patienten mit chronischer lymphatischer Leukämie (CLL) eingeführt worden, wo er sich als signifikant besser erweist als die Vorgängersubstanz Rituximab [1]. Aber auch jenseits der Immunologie gibt es Fortschritte: Der B-Zell-Rezeptor, dessen Aktivität essenziell für die Pathogenese der NHL ist, weist in seinem Signaltransduktionsweg einige interessante Zielstrukturen für neue Medikamente auf, und erstmals werden bei Lymphomen auch Substanzen erprobt, die die Apoptose-Hemmung in Tumorzellen aufheben können. Schließlich könnte der Proteasominhibitor Bortezomib, bisher nur beim multiplen Myelom zugelassen, künftig auch beim Mantelzell-Lymphom eine Rolle spielen.
Die CLL ist nach wie vor unheilbar – außer durch eine allogene Stammzelltransplantation, die aber bei den meist älteren Patienten nicht so häufig infrage kommt – und die Patienten entwickeln in den meisten Fällen früher oder später eine resistente bzw. refraktäre Erkrankung. Eine zentrale Schaltstelle im Signalweg des B-Zell-Rezeptors ist Bruton´s Tyrosinkinase (Btk), die sich mit dem spezifischen Inhibitor Ibrutinib sehr wirksam hemmen lässt, wie sich in frühen klinischen Studien bei verschiedenen Lymphomen bereits gezeigt hatte. Bei der EHA-Jahrestagung stellte Peter Hillmen, Leeds, die Ergebnisse der Phase-III-Studie RESONATE vor, in der 391 Patienten mit rezidivierter oder therapierefraktärer CLL oder „small lymphocytic lymphoma“ (SLL) und schlechter Prognose randomisiert entweder den CD20-Antikörper Ofatumumab oder Ibrutinib (420 mg/d bis zur Progression) erhielten [2].
Primärer Endpunkt der Studie, die inzwischen auch publiziert ist [3], war das progressionsfreie Überleben, das durch Ibrutinib gegenüber Ofatumumab signifikant verlängert wurde: Im Ibrutinib-Arm war der Medianwert noch nicht erreicht, unter Ofatumumab lag er bei 8,1 Monaten; die Hazard Ratio war mit 0,215 extrem niedrig, d. h. das Risiko für Krankheitsprogression oder Tod wurde um 78,5% reduziert (p < 0,0001). Sogar das Gesamtüberleben war nach einer medianen Behandlungsdauer von nur 9,4 Monaten unter Ibrutinib signifikant länger (HR 0,434; p = 0,0049), d. h. das Mortalitätsrisiko war um 57% reduziert. Auch Patienten mit Hochrisikoerkrankungen etwa aufgrund von zytogenetischen Risikofaktoren wie einer 17p-Deletion oder Refraktärität gegen Fludarabin sprachen auf Ibrutinib genauso an wie diejenigen ohne solche Risikofaktoren. Die häufigste Nebenwirkung von Ibrutinib war eine Diarrhö, die aber meist nur während der ersten Wochen auftrat und selbstlimitierend war, so Hillmen.
Nach diesen Ergebnissen wird Ibrutinib künftig sicherlich eine wichtige Rolle in der Therapie von rezidivierter/refraktärer CLL oder SLL spielen. In den USA wurde es bereits im Februar auf der Basis von Phase-II-Daten in einem beschleunigten Verfahren zugelassen, in Europa ist die Zulasssung beantragt.
PI3K-Inhibitor auch in Hochrisikogruppen hochwirksam
Seit Kurzem zugelassen (für die rezidivierte CLL und das refraktäre follikuläre Lymphom) ist Idelalisib, ein spezifischer Inhibitor der δ-Isoform der Phosphatidylinositol-3-Kinase (PI3Kδ). Aus der Zulassungsstudie für die CLL, die unter Beteiligung der Deutschen CLL-Studiengruppe durchgeführt wurde, stellte Steven Coutre, Stanford, die Ergebnisse der zweiten geplanten Interimanalyse vor [4]. 220 Patienten mit rezidivierter CLL und einem Allgemeinzustand, der eine zytotoxische Chemotherapie nicht gestattete, hatten Rituximab und dazu in doppelblinder Manier entweder Placebo oder zweimal täglich 150 mg Idelalisib erhalten. Patienten in der Placebogruppe konnten bei Progression ihrer Erkrankung ebenfalls den PI3Kδ-Inhibitor bekommen. Nachdem die Studie bereits nach der ersten Interimsanalyse wegen „überwältigender Wirksamkeit“ beendet worden war [5], bestätigte die zweite Analyse diese Ergebnisse:
Bei der progressionsfreien Überlebenszeit war der Medianwert in der Idelalisib-Gruppe noch nicht erreicht, unter Rituximab alleine betrug er 5,5 Monate (Hazard Ratio 0,18; p < 0,0001); nach 24 Wochen waren mit Idelalisib 90%, mit Placebo 50% der Patienten progressionsfrei am Leben [4]. Selbst beim Gesamtüberleben zeigte sich bereits nach so kurzer Zeit und trotz des Crossover-Designs bereits ein signifikanter Vorteil: Die Medianwerte sind in beiden Armen noch nicht erreicht, nach 24 Wochen waren unter Idelalisib noch 96%, unter Placebo 86% der Patienten am Leben (HR 0,28; p = 0,003).
Das Bemerkenswerteste an dieser Substanz ist vielleicht die gleichmäßige Wirksamkeit in allen untersuchten Subgruppen von Patienten, die Stephan Stilgenbauer, Ulm, deutlich machte [6]: Selbst in der Gruppe mit dem höchsten Risiko – denjenigen, die eine Deletion 17p bzw. eine TP53-Mutation aufwiesen – lag die Ansprechrate bei 76,5% und die Hazard Ratio für progressionsfreies Überleben bei 0,13, verglichen mit 80,4% und 0,17 bei denen ohne eine der beiden Alterationen. Die europäische Zulassung gilt deshalb für Patienten mit 17p-Deletion oder TP53-Mutation, die für eine Chemoimmuntherapie nicht geeignet sind, bereits in der Erstlinie. Die Wirksamkeit von Idelalisib ging mit einem sehr akzeptablen Sicherheitsprofil einher: Mit 56% lag der Anteil der Patienten mit Nebenwirkungen vom Grad 3 oder mehr kaum viel höher als in der Placebogruppe (48%).
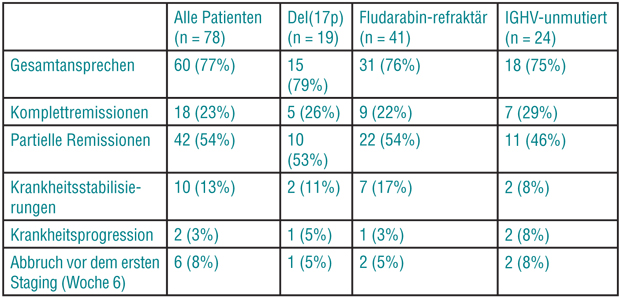
Apoptose-Inhibitor bei fortgeschrittener CLL
Ein ganz zentraler Mechanismus, mit dem die Homöostase aller Körperzellen geregelt wird, der aber in Tumorzellen häufig inaktiviert ist, ist die Apoptose. Sie kann durch das in der Mitochondrienmembran vorliegende Protein Bcl-2 unterdrückt werden, weshalb selektive, hochpotente und oral verfügbare Bcl-2-Inhibitoren wie ABT-199 entwickelt wurden. Bcl-2 ist in CLL-Zellen überexprimiert und führt zur Dysregulation des Apoptose-Mechanismus, sodass die leukämischen Zellen nicht nur überleben, sondern auch resistent gegen Zytostatika werden.
ABT-199 bewirkt in vitro sehr schnell und effizient die Apoptose von CLL-Zellen. In einer Phase-I-Studie konnte bei rezidivierter oder refraktärer CLL oder „small lymphocytic lymphoma“ (SLL) eine hohe Anti-Tumor-Wirksamkeit mit kompletten Remissionen beobachtet werden [7]. Die Substanz war so potent, dass es gelegentlich zu einem Tumor-Lyse-Syndrom kam; ein Patient verstarb daran. Nach Umstellung auf eine einschleichende Dosierung von anfangs 20 mg/d, die über fünf Wochen auf zuletzt 400 mg/d erhöht wird, so John Seymour, Adelaide, kam es zu keinem Tumor-Lyse-Syndrom mehr.
Die Wirksamkeit des Inhibitors bei den bisher 105 eingeschlossenen Patienten war beeindruckend (s. Tab.): Gut drei Viertel der Patienten sprachen auf ihn an, etwa ein Viertel mit einer Komplettremission. Vor allem aber scheint das Ansprechen auch hier völlig unabhängig von herkömmlichen Risikofaktoren wie del(17p), einer Refraktärität gegen Fludarabin oder unmutierten IGHV-Genen zu sein. Einige Patienten mit Komplettremission wurden auch MRD-negativ.
Die Verträglichkeit von ABT199 war gut: Am häufigsten waren Diarrhö (40%), Neutropenie (36%) und Nausea (35%) , aber die einzige Grad-3/4-Toxizität bei mehr als 10% der Patienten war eine Neutropenie (33%). Die Dosierung von 400 mg/d wird derzeit in einer erweiterten Sicherheitskohorte von Patienten untersucht. Der Medianwert des progressionsfreien Überlebens wurde bisher bei Dosen von 400 mg/d oder höher noch nicht erreicht, die geschätzte 2-Jahres-Rate liegt bei 59%. In einer Reihe klinischer Studien wird ABT-199 derzeit weiter untersucht – in einer Phase-II-Studie etwa in Monotherapie bei Patienten mit del(17p). In einer Phase-III-Studie wird es in Kombination mit Rituximab mit Bendamustin/Rituximab verglichen.
Mantelzell-Lymphom: neuer Standard
Die eher aggressiven Mantelzell-Lymphome machen etwa 6% der Non-Hodgkin-Lymphome aus. Im Gegensatz zum diffus-großzelligen B-Zell-Lymphom sind sie außer durch eine allogene Stammzelltransplantation nicht heilbar. Vor allem bei älteren Patienten, die auch keine autologe Stammzelltransplantation mehr bekommen können, ist bislang eine Immunchemotherapie mit R-CHOP (Rituximab, Cyclophosphamid, Doxorubicin, Vincristin und Prednison) der Standard in der Erstlinie – mit allerdings begrenztem progressionsfreiem Überleben. In einer internationalen Phase-III-Studie wurde R-CHOP daher mit einem Protokoll verglichen, in dem der Proteasominhibitor Bortezomib, der in den USA in der rezidivierten Situation zugelassen ist, anstelle von Vincristin eingesetzt wurde (VR-CAP, [8]).
In 128 Zentren in 28 Ländern Europas, Asiens und Südamerikas wurden 487 Patienten mit neu diagnostiziertem Mantelzell-Lymphom randomisiert, für die eine Stammzelltransplantation nicht infrage kam, so Tadeusz Robak, Lodz. Vorgesehen waren bis zu acht Therapiezyklen, in beiden Armen wurden median sechs Zyklen gegeben. Primärer Endpunkt war das mediane progressionsfreie Überleben, das durch VR-CAP um zehn Monate von 14,4 auf 24,7 Monate und damit um 59% signifikant verlängert wurde (HR 0,63; p < 0,001), und zwar in allen untersuchten Subgruppen. Die Gesamtansprechrate war in beiden Armen vergleichbar mit 92% vs. 90%, aber unter VR-CAP gab es mit 53% vs. 42% signifikant mehr Komplettremissionen als mit R-CHOP (p = 0,007). Signifikant überlegen war VR-CAP auch bei der medianen Dauer des Ansprechens (36,5 vs. 15,1 Monate) und bei der medianen Dauer der Komplettremissionen (42,1 vs. 18,0 Monate). Die Gesamtüberlebensraten unterschieden sich nach vier Jahren um etwa 10% zugunsten von VR-CAP (64,4% vs. 53,9%), eine Differenz, die allerdings noch nicht signifikant ist (HR 0,80; p = 0,173).
Thrombozytopenien vom Grad 3 oder höher waren unter VR-CAP zwar deutlich häufiger (57% vs. 6%), nicht aber Grad-3/4-Blutungen (1,7% vs. 1,2%). Weil Bortezomib ja das ebenfalls neurotoxische Vincristin ersetzt hatte, waren Grad-3/4-Neuropathien unter VR-CAP nicht wesentlich häufiger als unter R-CHOP (7,5% vs. 4,1%); sie dürften sich mit der neuen subkutanen Galenik des Proteasominhibitors noch stärker reduzieren lassen, so Robak. Seiner Meinung nach könnte VR-CAP sich mit diesen Resultaten als ein neuer Standard für Patienten mit neu diagnostiziertem Mantelzell-Lymphom, die für eine Hochdosistherapie nicht infrage kommt, etablieren.
Josef Gulden
Literatur:
1. Goede V et al. N Engl J Med 2014; 370: 1101-10.
2. Hillmen P et al. Haematologica 2014; 99 (s1): 244 (EHA 2014, Abstract #S693).
3. Byrd JC et al. N Engl J Med 2014; 371: 213-23.
4. Coutre SE et al. Haematologica 2014; 99 (s1): 249-50 (EHA 2014, Abstract #S704).
5. Furman RR et al. N Engl J Med 2014; 370: 997-1007.
6. Stilgenbauer S et al. Haematologica 2014; 99 (s1): 521 (EHA 2014, Abstract #S1341).
7. Seymour JF et al. Haematologica 2014; 99 (s1): 249 (EHA 2014, Abstract #S702).
8. Robak T et al. Haematologica 2014; 99 (s1): 523-4 (EHA 2014, Abstract #S1345).
19th Congress of the European Hematology Association (EHA), 12.-15.6.2014 in Mailand.