Molekularpathologie des Lungenkarzinoms
Das Lungenkarzinom ist weltweit immer noch die häufigste krebsassoziierte Todesursache bei Männern und die dritthäufigste bei Frauen. Von histopathologischer Seite werden die Lungentumoren in kleinzellige (SCLC) und nicht-kleinzellige Lungenkarzinome (NSCLC) unterteilt. Die sehr heterogene Gruppe der NSCLC wird weiter anhand histologischer Kriterien in die drei Typen Adenokarzinome, Plattenepithelkarzinome und großzellige Karzinome gegliedert. Die Adenokarzinome zeigen in der Regel einen drüsigen Aufbau mit teils azinärem, teils trabekulärem und teils auch solidem Wachstumsmuster. Demgegenüber imitieren die Plattenepithelkarzinome, wie der Name sagt, das Plattenepithel mit Ausbildung von Interzellular-Brücken und Verhornung. Die großzelligen Karzinome zeigen keine spezifische Differenzierung und können zum Teil nur mit immunhistochemischen Methoden von hochmalignen Non-Hodgkin-Lymphomen und Melanomen unterschieden werden. Sie können durch Ent-Differenzierung sowohl aus Adenokarzinomen als auch aus Plattenepithelkarzinomen entstehen und teilen daher auch molekulare Besonderheiten mit beiden Gruppen. Genetische Untersuchungen in den letzten Jahren haben allerdings demonstriert, dass der größere Teil der großzelligen Karzinome auf molekularer Ebene den Adenokarzinomen zuzuordnen ist [1].
In den letzten 15 Jahren hat das Wissen um die genetischen Veränderungen beim Lungenkrebs exponentiell zugenommen. Am besten ist hier das Adenokarzinom untersucht. Bei Nichtrauchern und hier vor allen Dingen bei jüngeren Frauen wurden Mutationen des Rezeptors für den epidermalen Wachstumsfaktor (EGFR) zunächst in asiatischen Patientenkollektiven am häufigsten beobachtet [2]. EGFR ist ein Rezeptor für Wachstumsfaktoren, der aktivierende Mutationen in den Exons 18, 19, 20 und 21 aufweisen kann. Die Mutationen in Exon 18, 19 und 21 sensitivieren diese Karzinome für EGFR-Tyrosinkinaseinhibitoren (EGFR-TKI [3]), während Mutationen in Exon 20 zu einer Resistenz führen. Mithilfe von EGFR-TKI lässt sich das progressionsfreie Überleben der betroffenen Patienten um durchschnittlich 10–16 Monate verlängern [3].
Die Pathogenese der Adenokarzinome basiert aber nur bei ca. 15% der betroffenen Europäer auf EGFR-Mutationen, sodass nur ein relativ geringer Prozentsatz der Patienten von dieser neuen Therapieoption profitiert. Die meisten dieser Patienten sind außerdem Nichtraucher. Hauptsächlich bei Nichtrauchern zeigt sich auch eine andere Mutation, die sehr erfolgversprechende therapeutische Optionen eröffnet, nämlich eine Translokation, die das ALK-Gen (Anaplastic Lymphoma Kinase) involviert. Hier sind ungefähr 7–8% aller Patienten mit Adenokarzinomen betroffen.
Translokationen des ALK-Gens sind auch aus anderen Tumorentitäten bekannt, aber beim NSCLC ist das Fusionsprodukt praktisch immer EML4-ALK [4]. Hier bieten ALK-Inhibitoren die Möglichkeit einer zielgerichteten Therapie, die ebenfalls eine Lebensverlängerung um durchschnittlich mehrere Monate bei geringen Nebenwirkungen ermöglicht [5]. Sowohl die Therapie mit EGFR-TKI als auch die mit ALK-Inhibitoren zeigen aber leider keinen langfristigen Erfolg. In beiden Fällen entwickeln sich im Durchschnitt nach ungefähr einem Jahr Resistenzen, die zu einer Progression des Karzinoms führen [6]. Hier werden derzeit Second-line-Therapien entwickelt, die diese resistenten Klone angreifen können.
Die pathogenetische Entwicklung von Adenokarzinomen bei Rauchern scheint generell eher andere Mechanismen zu involvieren. Vor allen Dingen tritt hier sehr häufig (ca. 30% aller Raucher) eine Mutation im KRAS-Gen (Kirsten rat sarcoma viral oncogene homolog) auf, die mit einer deutlich schlechteren Prognose korreliert [1]. Zusätzlich hat sich diese Mutation bisher allen pharmakologischen Angriffen erfolgreich widersetzt, sodass keine gezielte Therapie für Tumoren mit dieser Mutation existiert. KRAS ist ein Signalprotein im EGFR-Signalweg, welches diesen Signalweg unabhängig von EGFR-abhängigen Proliferationssignalen aktiviert. Damit sind EGFR-Inhibitoren bei dieser Mutation normalerweise wirkungslos [7]. Da sich KRAS selbst als pharmakologisches Target als schwer angreifbar erwiesen hat, wird derzeit versucht, die von KRAS ausgehenden proliferativen Signale bei nachfolgenden Signalmolekülen zu inhibieren. Hier kommen z. B. MEK-Inhibitoren ins Spiel, die derzeit in klinischen Studien getestet werden [8]. Da EGFR- und KRAS-Mutationen denselben Signalweg betreffen, schließen sie sich normalerweise gegenseitig aus.
Mit EGFR-, KRAS- und AKL-Mutationen lassen sich ca. die Hälfte aller Adenokarzinome molekulargenetisch definieren. Die Suche nach weiteren, pathogenetisch relevanten Mutationen hat Aberrationen in verschiedenen Genen an den Tag gebracht, die aber in jedem Fall weniger als 5% aller Patienten mit Adenokarzinomen betreffen [9].
Hier muss vor allem auch die ROS1-Translokation (1–3% aller Adenokarzinome) erwähnt werden, für die bereits eine zielgerichtete Therapie bekannt ist [10]. Es hat sich gezeigt, dass Patienten mit dieser Translokation auf die ALK-Inhibitoren ansprechen, die ursprünglich für Tumoren mit ALK-Translokationen entwickelt worden waren. Auch hier ist der Therapieerfolg transient, d. h. nach ca. einem Jahr entwickeln sich Resistenzen. Eine weitere Parallele zu den Patienten mit ALK-Translokationen besteht darin, dass es sich auch bei diesen ROS1-translozierten Patienten überwiegend um Nichtraucher handelt.
Ganz anders steht es mit Mutationen, die das BRAF-Gen betreffen, dessen Proteinprodukt ein weiteres Signal-Molekül im Signalweg diverser Wachstumsfaktoren darstellt. Diese Mutation ist auch von kolorektalen Karzinomen bekannt, wo sie mit einer infausten Prognose einhergeht [14]. Auch beim NSCLC gibt es Hinweise, dass diese Mutation mit einer sehr schlechten Prognose verbunden ist [15]. Hier sind allerdings nur ca. 1–3% aller NSCLC-Patienten von BRAF-Mutationen betroffen, und zwar sowohl Patienten mit Adenokarzinomen als auch mit Plattenepithelkarzinomen. Die BRAFV600E-Mutation kommt auch in ca. der Hälfte aller Melanome vor, wo sie sehr erfolgreich mit BRAF-Inhibitoren behandelt wird [16]. Leider verläuft der Signalweg in Melanomen aber anders (über den Rezeptor KIT) als in NSCLC und kolorektalen Karzinomen (über EGFR). Es hat sich gezeigt, dass die bisher bekannten BRAF-Inhibitoren in diesen Adenokarzinomen eher sogar eine adverse Wirkung zeigen, sodass eine zielgerichtete Therapie für diese aggressiven Tumoren bisher nicht bekannt ist [17].
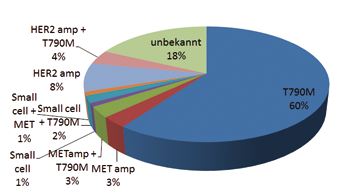
Sowohl die Therapie mit EGFR-TKI als auch die mit ALK-Inhibitoren zeigt aber leider keinen langfristigen Erfolg. In beiden Fällen entwickeln sich im Durchschnitt nach ungefähr einem Jahr Resistenzen, die zu einer Progression des Karzinoms führen [6]. Die häufigsten Resistenzmechanismen sind Mutationen in der Kinase-Domäne der betreffenden Gene, die die Bindung der entsprechenden Inhibitoren blockieren. Im EGFR-Gen tritt zum Beispiel die T790M-Mutation auf, die sich bei über 50% aller EGFR-TKI-resistenten Tumoren finden lässt. Entsprechende Mutationen sind inzwischen auch in der Kinase-Domäne von ALK gefunden worden. Derzeit werden Second-line-Therapien entwickelt, die diese resistenten Klone angreifen können. In ca. 5–10% aller TKI-resistenten Tumoren treten auch Amplifikationen des MET-Gens auf.
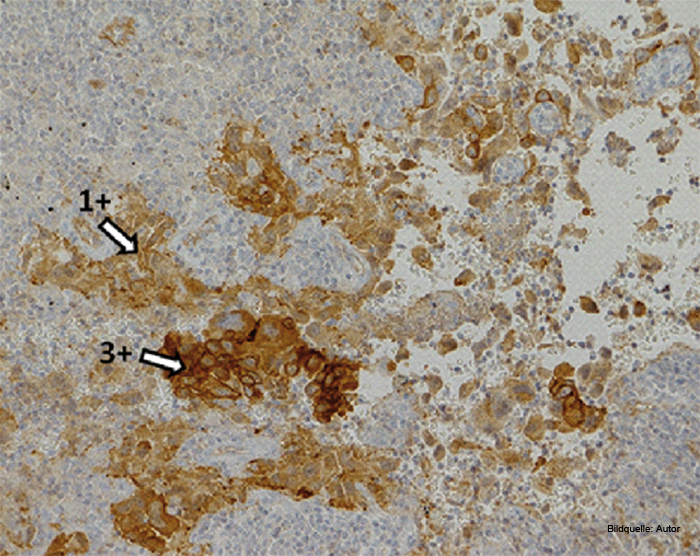
Die MET-Amplifikation ist eine weitere Mutation, die die Grenzen von Adeno- und Plattenepithelkarzinomen überschreitet. Sie findet sich in ungefähr 5% aller NSCLC als primäre Aberration [19]. Auch Punktmutationen des MET-Gens kommen vor, sind aber sehr selten [20]. MET ist wie EGFR ein Rezeptor für Wachstumsfaktoren, speziell für den „Hepatocyte Growth Factor". Da er sich auf der Zelloberfläche befindet, bietet er eine Angriffsfläche für den therapeutischen Einsatz von spezifischen Antikörpern, die sich zum Teil bereits in Phase-III-Studien befinden. Als Nachweistechniken kommen hier FISH (Fluoreszenz-in-situ-Hybridisierung) und Immunhistochemie in Betracht. Die Immunhistochemie hat sich dabei als gleichwertig in der Spezifität und als deutlich robuster und kostengünstiger erwiesen. Die Interpretation ist allerdings nicht trivial, sodass eine spezifische Schulung der Pathologen (ähnlich wie bei der Immunhistochemie für HER2) erfolgen sollte. Des Weiteren sollten nur validierte Antikörper und Detektionssysteme verwendet werden.
Weitere Mutationen bei Adenokarzinomen betreffen z. B. die KRAS-Verwandten HRAS und NRAS, die Rezeptoren HER2 und RET, sowie das Signalmolekül PIK3CA 9,21. Da alle diese Mutationen sehr selten auftreten, sind zielgerichtete Therapien nur für diejenigen von ihnen bekannt, die auch bei anderen Tumoren eine Rolle spielen und bei denen die Therapieoptionen sich auf die Entität der NSCLC übertragen ließen (z. B. PIK3CA-Mutationen und mTOR-Inhibitoren). Andere Mutationen sind bei einzelnen Patienten gefunden worden, aber mehr als ein Drittel der Adenokarzinome können immer noch nicht molekulargenetisch charakterisiert werden. Dies betrifft vor allem die Pathogenese der Rauch-induzierten Karzinome.
Die molekulare Pathogenese der Plattenepithelkarzinome hat sich generell als weniger leicht zugänglich erwiesen, obwohl hier in den letzten Jahren erheblich Fortschritte gemacht wurden. Circa ein Drittel dieser Karzinome zeigen Amplifikationen des PIK3CA-Gens [12]. Hier wird es nötig sein, die molekularen Folgen einer PIK3CA-Amplifikation genauer zu definieren, um zielgerichtet eingreifen zu können.
Eine bessere therapeutische Angriffsfläche bietet die Amplifikation des FGFR1 (fibroblast growth factor receptor 1). Hier wurde bereits ein FGFR-spezifischer TKI entwickelt, der jetzt in klinischen Studien getestet wird [22]. Die hier betroffenen 20-30% der Patienten mit Plattenepithelkarzinomen sollten daher hoffentlich bald von einer zielgerichteten Therapie profitieren können.
Eine Mutation, die spezifisch ist für Plattenepithelkarzinome und die sich heute möglicherweise bereits zielgerichtet angreifen lässt, betrifft das DDR2 Gen. Tumoren mit dieser Mutation können auf eine Therapie mit einem ABL-Inhibitor ansprechen [23]. Dieser TKI wurde ursprünglich als Alternative zu den first line TKI bei der chronischen myeloischen Leukämie entwickelt, zeigt aber durchaus umfassenderes therapeutisches Potenzial. Hier allerdings stößt man diagnostisch auf Probleme. Im Gegensatz zu den Mutationen in EGFR, KRAS oder auch BRAF und PIK3CA, die sich auf wenige Regionen des Gens beschränken, können Mutationen in DDR2 sich über das ganze, sehr große Gen erstrecken. Dies macht es erforderlich, das gesamte Gen zu sequenzieren, um zu einer therapeutischen Entscheidung zu kommen. Hier wird es nötig werden, die neue Technik des Next Generation Sequencing (NGS) in die Diagnostik zu integrieren, die eine Sequenzierung großer Gene erheblich schneller (und kostengünstiger) macht. Das NGS ermöglicht es, dem Patienten und dem behandelnden Arzt die Informationen zum Mutationsstatus in kurzer Zeit zur Verfügung zu stellen, um ein rechtzeitiges Einleiten der Therapie zu ermöglichen. Auch bei den Plattenepithelkarzinomen wurden aber für ungefähr ein Drittel der Tumoren noch keine pathogenetisch relevante Mutation gefunden, sodass die Möglichkeiten einer zielgerichteten Therapie hier noch erheblich eingeschränkter sind als bei den Adenokarzinomen.
Die letzte, bislang unerwähnte Gruppe der kleinzelligen Lungenkarzinome bildet eine vergleichsweise seltene Tumorentität, die stark mit Tabakkonsum assoziiert ist und für die bisher nur wenige potenzielle Targets beschrieben sind. In jüngerer Zeit wurden nun aber auch für diese klinisch schwierige Entität einige genetische Aberrationen beschrieben, die Hoffnung auf neue therapeutische Optionen machen. In 6% der Fälle wurden FGFR1-Amplifikationen beschrieben [13]. Diese haben sich beim Plattenepithelkarzinom (hier in 22% der Fälle) bereits als sensitiv auf FGFR-Inhibition erwiesen [22].
Bei weiteren 10% liegen inaktivierende Mutationen in der Phosphatase PTEN vor. PTEN ist ein negativer Regulator des PI3-Kinase-Signalwegs, dessen Mutationen in der Regel zum Funktionsverlust des Proteins führen. Dies hat zur Folge, dass der PI3-Kinase Signalweg überaktiviert wird, womit sich die Möglichkeit für einen zielgerichteten Therapieansatz eröffnet. Abgesehen von diesen Anfangserfolgen ist der Großteil der pathogenetisch relevanten Mutationen beim kleinzelligen Karzinom jedoch noch unbekannt. Das kleinzellige Lungenkarzinom bleibt daher weiterhin eine klinisch schwierige Gruppe mit äußerst ungünstiger Prognose.
Mit der zunehmenden Kenntnis der genetischen Grundlagen der Lungenkarzinome wird eine immer umfassendere und sensitivere molekulare Diagnostik nötig, die möglichst auch von kleinen Biopsien und zytologischen Präparaten zuverlässige Ergebnisse liefert. Ein wesentliches Hilfsmittel ist hier die Mikrodissektion (manuell oder – noch besser – lasergesteuert), die es ermöglicht, Tumorzellen aus dem umgebenden Normalgewebe zu isolieren und gezielt zu analysieren. Sensitivere Methoden, wie Realtime-PCR-basierte Ansätze und Next Generation Sequencing (NGS), ermöglichen die Detektion von Mutationen auch in Subklonen des Tumors oder bei einem insgesamt geringen Tumoranteil.
Ein weiterer Vorteil des NGS ist es auch, dass viele Gene gleichzeitig und kostengünstig evaluiert werden können. Da viele der neu entdeckten Mutationen nur in einem kleinen Prozentsatz der Patienten gefunden worden sind, ist nur bei gleichzeitiger Testung aller dieser Gene in angemessen kurzer Zeit auch die molekulare Diagnose für den Patienten verfügbar. Neue Techniken, die derzeit entwickelt werden, werden es auch ermöglichen, Punktmutationen, Amplifikationen und Translokationen in einem Ansatz zu untersuchen. Dies lässt hoffen, dass in Zukunft komplexe genetische Analysen des Genoms oder des Exoms helfen werden, auch die bisher unbekannten Mechanismen der Karzinomentstehung und Progression zu ermitteln und ggf. therapeutisch anzugreifen.
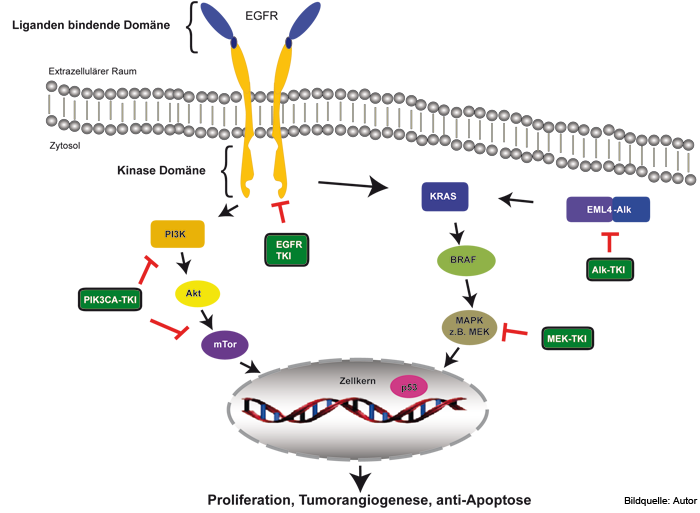
Abb. 1: EGFR-Signalweg und therapeutische Ansatzpunkte.
Mit EGFR-, KRAS- und AKL-Mutationen lassen sich ca. die Hälfte aller Adenokarzinome molekulargenetisch definieren. Die Suche nach weiteren, pathogenetisch relevanten Mutationen hat Aberrationen in verschiedenen Genen an den Tag gebracht, die aber in jedem Fall weniger als 5% aller Patienten mit Adenokarzinomen betreffen [9].
Hier muss vor allem auch die ROS1-Translokation (1–3% aller Adenokarzinome) erwähnt werden, für die bereits eine zielgerichtete Therapie bekannt ist [10]. Es hat sich gezeigt, dass Patienten mit dieser Translokation auf die ALK-Inhibitoren ansprechen, die ursprünglich für Tumoren mit ALK-Translokationen entwickelt worden waren. Auch hier ist der Therapieerfolg transient, d. h. nach ca. einem Jahr entwickeln sich Resistenzen. Eine weitere Parallele zu den Patienten mit ALK-Translokationen besteht darin, dass es sich auch bei diesen ROS1-translozierten Patienten überwiegend um Nichtraucher handelt.
Ganz anders steht es mit Mutationen, die das BRAF-Gen betreffen, dessen Proteinprodukt ein weiteres Signal-Molekül im Signalweg diverser Wachstumsfaktoren darstellt. Diese Mutation ist auch von kolorektalen Karzinomen bekannt, wo sie mit einer infausten Prognose einhergeht [14]. Auch beim NSCLC gibt es Hinweise, dass diese Mutation mit einer sehr schlechten Prognose verbunden ist [15]. Hier sind allerdings nur ca. 1–3% aller NSCLC-Patienten von BRAF-Mutationen betroffen, und zwar sowohl Patienten mit Adenokarzinomen als auch mit Plattenepithelkarzinomen. Die BRAFV600E-Mutation kommt auch in ca. der Hälfte aller Melanome vor, wo sie sehr erfolgreich mit BRAF-Inhibitoren behandelt wird [16]. Leider verläuft der Signalweg in Melanomen aber anders (über den Rezeptor KIT) als in NSCLC und kolorektalen Karzinomen (über EGFR). Es hat sich gezeigt, dass die bisher bekannten BRAF-Inhibitoren in diesen Adenokarzinomen eher sogar eine adverse Wirkung zeigen, sodass eine zielgerichtete Therapie für diese aggressiven Tumoren bisher nicht bekannt ist [17].
Sowohl die Therapie mit EGFR-TKI als auch die mit ALK-Inhibitoren zeigt aber leider keinen langfristigen Erfolg. In beiden Fällen entwickeln sich im Durchschnitt nach ungefähr einem Jahr Resistenzen, die zu einer Progression des Karzinoms führen [6]. Die häufigsten Resistenzmechanismen sind Mutationen in der Kinase-Domäne der betreffenden Gene, die die Bindung der entsprechenden Inhibitoren blockieren. Im EGFR-Gen tritt zum Beispiel die T790M-Mutation auf, die sich bei über 50% aller EGFR-TKI-resistenten Tumoren finden lässt. Entsprechende Mutationen sind inzwischen auch in der Kinase-Domäne von ALK gefunden worden. Derzeit werden Second-line-Therapien entwickelt, die diese resistenten Klone angreifen können. In ca. 5–10% aller TKI-resistenten Tumoren treten auch Amplifikationen des MET-Gens auf.
Die MET-Amplifikation ist eine weitere Mutation, die die Grenzen von Adeno- und Plattenepithelkarzinomen überschreitet. Sie findet sich in ungefähr 5% aller NSCLC als primäre Aberration [19]. Auch Punktmutationen des MET-Gens kommen vor, sind aber sehr selten [20]. MET ist wie EGFR ein Rezeptor für Wachstumsfaktoren, speziell für den „Hepatocyte Growth Factor". Da er sich auf der Zelloberfläche befindet, bietet er eine Angriffsfläche für den therapeutischen Einsatz von spezifischen Antikörpern, die sich zum Teil bereits in Phase-III-Studien befinden. Als Nachweistechniken kommen hier FISH (Fluoreszenz-in-situ-Hybridisierung) und Immunhistochemie in Betracht. Die Immunhistochemie hat sich dabei als gleichwertig in der Spezifität und als deutlich robuster und kostengünstiger erwiesen. Die Interpretation ist allerdings nicht trivial, sodass eine spezifische Schulung der Pathologen (ähnlich wie bei der Immunhistochemie für HER2) erfolgen sollte. Des Weiteren sollten nur validierte Antikörper und Detektionssysteme verwendet werden.
Weitere Mutationen bei Adenokarzinomen betreffen z. B. die KRAS-Verwandten HRAS und NRAS, die Rezeptoren HER2 und RET, sowie das Signalmolekül PIK3CA 9,21. Da alle diese Mutationen sehr selten auftreten, sind zielgerichtete Therapien nur für diejenigen von ihnen bekannt, die auch bei anderen Tumoren eine Rolle spielen und bei denen die Therapieoptionen sich auf die Entität der NSCLC übertragen ließen (z. B. PIK3CA-Mutationen und mTOR-Inhibitoren). Andere Mutationen sind bei einzelnen Patienten gefunden worden, aber mehr als ein Drittel der Adenokarzinome können immer noch nicht molekulargenetisch charakterisiert werden. Dies betrifft vor allem die Pathogenese der Rauch-induzierten Karzinome.
Die molekulare Pathogenese der Plattenepithelkarzinome hat sich generell als weniger leicht zugänglich erwiesen, obwohl hier in den letzten Jahren erheblich Fortschritte gemacht wurden. Circa ein Drittel dieser Karzinome zeigen Amplifikationen des PIK3CA-Gens [12]. Hier wird es nötig sein, die molekularen Folgen einer PIK3CA-Amplifikation genauer zu definieren, um zielgerichtet eingreifen zu können.
Eine bessere therapeutische Angriffsfläche bietet die Amplifikation des FGFR1 (fibroblast growth factor receptor 1). Hier wurde bereits ein FGFR-spezifischer TKI entwickelt, der jetzt in klinischen Studien getestet wird [22]. Die hier betroffenen 20-30% der Patienten mit Plattenepithelkarzinomen sollten daher hoffentlich bald von einer zielgerichteten Therapie profitieren können.
Eine Mutation, die spezifisch ist für Plattenepithelkarzinome und die sich heute möglicherweise bereits zielgerichtet angreifen lässt, betrifft das DDR2 Gen. Tumoren mit dieser Mutation können auf eine Therapie mit einem ABL-Inhibitor ansprechen [23]. Dieser TKI wurde ursprünglich als Alternative zu den first line TKI bei der chronischen myeloischen Leukämie entwickelt, zeigt aber durchaus umfassenderes therapeutisches Potenzial. Hier allerdings stößt man diagnostisch auf Probleme. Im Gegensatz zu den Mutationen in EGFR, KRAS oder auch BRAF und PIK3CA, die sich auf wenige Regionen des Gens beschränken, können Mutationen in DDR2 sich über das ganze, sehr große Gen erstrecken. Dies macht es erforderlich, das gesamte Gen zu sequenzieren, um zu einer therapeutischen Entscheidung zu kommen. Hier wird es nötig werden, die neue Technik des Next Generation Sequencing (NGS) in die Diagnostik zu integrieren, die eine Sequenzierung großer Gene erheblich schneller (und kostengünstiger) macht. Das NGS ermöglicht es, dem Patienten und dem behandelnden Arzt die Informationen zum Mutationsstatus in kurzer Zeit zur Verfügung zu stellen, um ein rechtzeitiges Einleiten der Therapie zu ermöglichen. Auch bei den Plattenepithelkarzinomen wurden aber für ungefähr ein Drittel der Tumoren noch keine pathogenetisch relevante Mutation gefunden, sodass die Möglichkeiten einer zielgerichteten Therapie hier noch erheblich eingeschränkter sind als bei den Adenokarzinomen.
Die letzte, bislang unerwähnte Gruppe der kleinzelligen Lungenkarzinome bildet eine vergleichsweise seltene Tumorentität, die stark mit Tabakkonsum assoziiert ist und für die bisher nur wenige potenzielle Targets beschrieben sind. In jüngerer Zeit wurden nun aber auch für diese klinisch schwierige Entität einige genetische Aberrationen beschrieben, die Hoffnung auf neue therapeutische Optionen machen. In 6% der Fälle wurden FGFR1-Amplifikationen beschrieben [13]. Diese haben sich beim Plattenepithelkarzinom (hier in 22% der Fälle) bereits als sensitiv auf FGFR-Inhibition erwiesen [22].
Bei weiteren 10% liegen inaktivierende Mutationen in der Phosphatase PTEN vor. PTEN ist ein negativer Regulator des PI3-Kinase-Signalwegs, dessen Mutationen in der Regel zum Funktionsverlust des Proteins führen. Dies hat zur Folge, dass der PI3-Kinase Signalweg überaktiviert wird, womit sich die Möglichkeit für einen zielgerichteten Therapieansatz eröffnet. Abgesehen von diesen Anfangserfolgen ist der Großteil der pathogenetisch relevanten Mutationen beim kleinzelligen Karzinom jedoch noch unbekannt. Das kleinzellige Lungenkarzinom bleibt daher weiterhin eine klinisch schwierige Gruppe mit äußerst ungünstiger Prognose.
Mit der zunehmenden Kenntnis der genetischen Grundlagen der Lungenkarzinome wird eine immer umfassendere und sensitivere molekulare Diagnostik nötig, die möglichst auch von kleinen Biopsien und zytologischen Präparaten zuverlässige Ergebnisse liefert. Ein wesentliches Hilfsmittel ist hier die Mikrodissektion (manuell oder – noch besser – lasergesteuert), die es ermöglicht, Tumorzellen aus dem umgebenden Normalgewebe zu isolieren und gezielt zu analysieren. Sensitivere Methoden, wie Realtime-PCR-basierte Ansätze und Next Generation Sequencing (NGS), ermöglichen die Detektion von Mutationen auch in Subklonen des Tumors oder bei einem insgesamt geringen Tumoranteil.
Ein weiterer Vorteil des NGS ist es auch, dass viele Gene gleichzeitig und kostengünstig evaluiert werden können. Da viele der neu entdeckten Mutationen nur in einem kleinen Prozentsatz der Patienten gefunden worden sind, ist nur bei gleichzeitiger Testung aller dieser Gene in angemessen kurzer Zeit auch die molekulare Diagnose für den Patienten verfügbar. Neue Techniken, die derzeit entwickelt werden, werden es auch ermöglichen, Punktmutationen, Amplifikationen und Translokationen in einem Ansatz zu untersuchen. Dies lässt hoffen, dass in Zukunft komplexe genetische Analysen des Genoms oder des Exoms helfen werden, auch die bisher unbekannten Mechanismen der Karzinomentstehung und Progression zu ermitteln und ggf. therapeutisch anzugreifen.
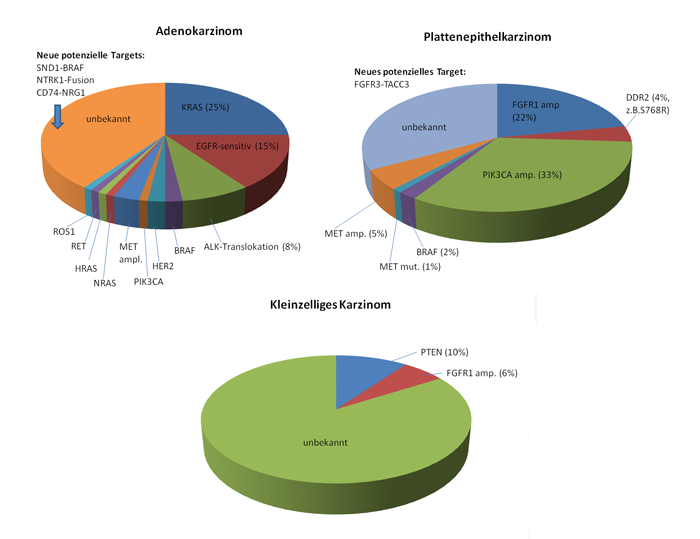
Abb. 2: Aktueller Stand der bekannten Treibermutationen des Bronchialkarzinoms [9,11,12,13].
Bildquelle: Autor
Ganz anders steht es mit Mutationen, die das BRAF-Gen betreffen, dessen Proteinprodukt ein weiteres Signal-Molekül im Signalweg diverser Wachstumsfaktoren darstellt. Diese Mutation ist auch von kolorektalen Karzinomen bekannt, wo sie mit einer infausten Prognose einhergeht [14]. Auch beim NSCLC gibt es Hinweise, dass diese Mutation mit einer sehr schlechten Prognose verbunden ist [15]. Hier sind allerdings nur ca. 1–3% aller NSCLC-Patienten von BRAF-Mutationen betroffen, und zwar sowohl Patienten mit Adenokarzinomen als auch mit Plattenepithelkarzinomen. Die BRAFV600E-Mutation kommt auch in ca. der Hälfte aller Melanome vor, wo sie sehr erfolgreich mit BRAF-Inhibitoren behandelt wird [16]. Leider verläuft der Signalweg in Melanomen aber anders (über den Rezeptor KIT) als in NSCLC und kolorektalen Karzinomen (über EGFR). Es hat sich gezeigt, dass die bisher bekannten BRAF-Inhibitoren in diesen Adenokarzinomen eher sogar eine adverse Wirkung zeigen, sodass eine zielgerichtete Therapie für diese aggressiven Tumoren bisher nicht bekannt ist [17].
Sowohl die Therapie mit EGFR-TKI als auch die mit ALK-Inhibitoren zeigt aber leider keinen langfristigen Erfolg. In beiden Fällen entwickeln sich im Durchschnitt nach ungefähr einem Jahr Resistenzen, die zu einer Progression des Karzinoms führen [6]. Die häufigsten Resistenzmechanismen sind Mutationen in der Kinase-Domäne der betreffenden Gene, die die Bindung der entsprechenden Inhibitoren blockieren. Im EGFR-Gen tritt zum Beispiel die T790M-Mutation auf, die sich bei über 50% aller EGFR-TKI-resistenten Tumoren finden lässt. Entsprechende Mutationen sind inzwischen auch in der Kinase-Domäne von ALK gefunden worden. Derzeit werden Second-line-Therapien entwickelt, die diese resistenten Klone angreifen können. In ca. 5–10% aller TKI-resistenten Tumoren treten auch Amplifikationen des MET-Gens auf.
Die MET-Amplifikation ist eine weitere Mutation, die die Grenzen von Adeno- und Plattenepithelkarzinomen überschreitet. Sie findet sich in ungefähr 5% aller NSCLC als primäre Aberration [19]. Auch Punktmutationen des MET-Gens kommen vor, sind aber sehr selten [20]. MET ist wie EGFR ein Rezeptor für Wachstumsfaktoren, speziell für den „Hepatocyte Growth Factor". Da er sich auf der Zelloberfläche befindet, bietet er eine Angriffsfläche für den therapeutischen Einsatz von spezifischen Antikörpern, die sich zum Teil bereits in Phase-III-Studien befinden. Als Nachweistechniken kommen hier FISH (Fluoreszenz-in-situ-Hybridisierung) und Immunhistochemie in Betracht. Die Immunhistochemie hat sich dabei als gleichwertig in der Spezifität und als deutlich robuster und kostengünstiger erwiesen. Die Interpretation ist allerdings nicht trivial, sodass eine spezifische Schulung der Pathologen (ähnlich wie bei der Immunhistochemie für HER2) erfolgen sollte. Des Weiteren sollten nur validierte Antikörper und Detektionssysteme verwendet werden.
Weitere Mutationen bei Adenokarzinomen betreffen z. B. die KRAS-Verwandten HRAS und NRAS, die Rezeptoren HER2 und RET, sowie das Signalmolekül PIK3CA 9,21. Da alle diese Mutationen sehr selten auftreten, sind zielgerichtete Therapien nur für diejenigen von ihnen bekannt, die auch bei anderen Tumoren eine Rolle spielen und bei denen die Therapieoptionen sich auf die Entität der NSCLC übertragen ließen (z. B. PIK3CA-Mutationen und mTOR-Inhibitoren). Andere Mutationen sind bei einzelnen Patienten gefunden worden, aber mehr als ein Drittel der Adenokarzinome können immer noch nicht molekulargenetisch charakterisiert werden. Dies betrifft vor allem die Pathogenese der Rauch-induzierten Karzinome.
Die molekulare Pathogenese der Plattenepithelkarzinome hat sich generell als weniger leicht zugänglich erwiesen, obwohl hier in den letzten Jahren erheblich Fortschritte gemacht wurden. Circa ein Drittel dieser Karzinome zeigen Amplifikationen des PIK3CA-Gens [12]. Hier wird es nötig sein, die molekularen Folgen einer PIK3CA-Amplifikation genauer zu definieren, um zielgerichtet eingreifen zu können.
Eine bessere therapeutische Angriffsfläche bietet die Amplifikation des FGFR1 (fibroblast growth factor receptor 1). Hier wurde bereits ein FGFR-spezifischer TKI entwickelt, der jetzt in klinischen Studien getestet wird [22]. Die hier betroffenen 20-30% der Patienten mit Plattenepithelkarzinomen sollten daher hoffentlich bald von einer zielgerichteten Therapie profitieren können.
Eine Mutation, die spezifisch ist für Plattenepithelkarzinome und die sich heute möglicherweise bereits zielgerichtet angreifen lässt, betrifft das DDR2 Gen. Tumoren mit dieser Mutation können auf eine Therapie mit einem ABL-Inhibitor ansprechen [23]. Dieser TKI wurde ursprünglich als Alternative zu den first line TKI bei der chronischen myeloischen Leukämie entwickelt, zeigt aber durchaus umfassenderes therapeutisches Potenzial. Hier allerdings stößt man diagnostisch auf Probleme. Im Gegensatz zu den Mutationen in EGFR, KRAS oder auch BRAF und PIK3CA, die sich auf wenige Regionen des Gens beschränken, können Mutationen in DDR2 sich über das ganze, sehr große Gen erstrecken. Dies macht es erforderlich, das gesamte Gen zu sequenzieren, um zu einer therapeutischen Entscheidung zu kommen. Hier wird es nötig werden, die neue Technik des Next Generation Sequencing (NGS) in die Diagnostik zu integrieren, die eine Sequenzierung großer Gene erheblich schneller (und kostengünstiger) macht. Das NGS ermöglicht es, dem Patienten und dem behandelnden Arzt die Informationen zum Mutationsstatus in kurzer Zeit zur Verfügung zu stellen, um ein rechtzeitiges Einleiten der Therapie zu ermöglichen. Auch bei den Plattenepithelkarzinomen wurden aber für ungefähr ein Drittel der Tumoren noch keine pathogenetisch relevante Mutation gefunden, sodass die Möglichkeiten einer zielgerichteten Therapie hier noch erheblich eingeschränkter sind als bei den Adenokarzinomen.
Die letzte, bislang unerwähnte Gruppe der kleinzelligen Lungenkarzinome bildet eine vergleichsweise seltene Tumorentität, die stark mit Tabakkonsum assoziiert ist und für die bisher nur wenige potenzielle Targets beschrieben sind. In jüngerer Zeit wurden nun aber auch für diese klinisch schwierige Entität einige genetische Aberrationen beschrieben, die Hoffnung auf neue therapeutische Optionen machen. In 6% der Fälle wurden FGFR1-Amplifikationen beschrieben [13]. Diese haben sich beim Plattenepithelkarzinom (hier in 22% der Fälle) bereits als sensitiv auf FGFR-Inhibition erwiesen [22].
Bei weiteren 10% liegen inaktivierende Mutationen in der Phosphatase PTEN vor. PTEN ist ein negativer Regulator des PI3-Kinase-Signalwegs, dessen Mutationen in der Regel zum Funktionsverlust des Proteins führen. Dies hat zur Folge, dass der PI3-Kinase Signalweg überaktiviert wird, womit sich die Möglichkeit für einen zielgerichteten Therapieansatz eröffnet. Abgesehen von diesen Anfangserfolgen ist der Großteil der pathogenetisch relevanten Mutationen beim kleinzelligen Karzinom jedoch noch unbekannt. Das kleinzellige Lungenkarzinom bleibt daher weiterhin eine klinisch schwierige Gruppe mit äußerst ungünstiger Prognose.
Mit der zunehmenden Kenntnis der genetischen Grundlagen der Lungenkarzinome wird eine immer umfassendere und sensitivere molekulare Diagnostik nötig, die möglichst auch von kleinen Biopsien und zytologischen Präparaten zuverlässige Ergebnisse liefert. Ein wesentliches Hilfsmittel ist hier die Mikrodissektion (manuell oder – noch besser – lasergesteuert), die es ermöglicht, Tumorzellen aus dem umgebenden Normalgewebe zu isolieren und gezielt zu analysieren. Sensitivere Methoden, wie Realtime-PCR-basierte Ansätze und Next Generation Sequencing (NGS), ermöglichen die Detektion von Mutationen auch in Subklonen des Tumors oder bei einem insgesamt geringen Tumoranteil.
Ein weiterer Vorteil des NGS ist es auch, dass viele Gene gleichzeitig und kostengünstig evaluiert werden können. Da viele der neu entdeckten Mutationen nur in einem kleinen Prozentsatz der Patienten gefunden worden sind, ist nur bei gleichzeitiger Testung aller dieser Gene in angemessen kurzer Zeit auch die molekulare Diagnose für den Patienten verfügbar. Neue Techniken, die derzeit entwickelt werden, werden es auch ermöglichen, Punktmutationen, Amplifikationen und Translokationen in einem Ansatz zu untersuchen. Dies lässt hoffen, dass in Zukunft komplexe genetische Analysen des Genoms oder des Exoms helfen werden, auch die bisher unbekannten Mechanismen der Karzinomentstehung und Progression zu ermitteln und ggf. therapeutisch anzugreifen.
Die MET-Amplifikation ist eine weitere Mutation, die die Grenzen von Adeno- und Plattenepithelkarzinomen überschreitet. Sie findet sich in ungefähr 5% aller NSCLC als primäre Aberration [19]. Auch Punktmutationen des MET-Gens kommen vor, sind aber sehr selten [20]. MET ist wie EGFR ein Rezeptor für Wachstumsfaktoren, speziell für den „Hepatocyte Growth Factor". Da er sich auf der Zelloberfläche befindet, bietet er eine Angriffsfläche für den therapeutischen Einsatz von spezifischen Antikörpern, die sich zum Teil bereits in Phase-III-Studien befinden. Als Nachweistechniken kommen hier FISH (Fluoreszenz-in-situ-Hybridisierung) und Immunhistochemie in Betracht. Die Immunhistochemie hat sich dabei als gleichwertig in der Spezifität und als deutlich robuster und kostengünstiger erwiesen. Die Interpretation ist allerdings nicht trivial, sodass eine spezifische Schulung der Pathologen (ähnlich wie bei der Immunhistochemie für HER2) erfolgen sollte. Des Weiteren sollten nur validierte Antikörper und Detektionssysteme verwendet werden.
Weitere Mutationen bei Adenokarzinomen betreffen z. B. die KRAS-Verwandten HRAS und NRAS, die Rezeptoren HER2 und RET, sowie das Signalmolekül PIK3CA 9,21. Da alle diese Mutationen sehr selten auftreten, sind zielgerichtete Therapien nur für diejenigen von ihnen bekannt, die auch bei anderen Tumoren eine Rolle spielen und bei denen die Therapieoptionen sich auf die Entität der NSCLC übertragen ließen (z. B. PIK3CA-Mutationen und mTOR-Inhibitoren). Andere Mutationen sind bei einzelnen Patienten gefunden worden, aber mehr als ein Drittel der Adenokarzinome können immer noch nicht molekulargenetisch charakterisiert werden. Dies betrifft vor allem die Pathogenese der Rauch-induzierten Karzinome.
Die molekulare Pathogenese der Plattenepithelkarzinome hat sich generell als weniger leicht zugänglich erwiesen, obwohl hier in den letzten Jahren erheblich Fortschritte gemacht wurden. Circa ein Drittel dieser Karzinome zeigen Amplifikationen des PIK3CA-Gens [12]. Hier wird es nötig sein, die molekularen Folgen einer PIK3CA-Amplifikation genauer zu definieren, um zielgerichtet eingreifen zu können.
Eine bessere therapeutische Angriffsfläche bietet die Amplifikation des FGFR1 (fibroblast growth factor receptor 1). Hier wurde bereits ein FGFR-spezifischer TKI entwickelt, der jetzt in klinischen Studien getestet wird [22]. Die hier betroffenen 20-30% der Patienten mit Plattenepithelkarzinomen sollten daher hoffentlich bald von einer zielgerichteten Therapie profitieren können.
Eine Mutation, die spezifisch ist für Plattenepithelkarzinome und die sich heute möglicherweise bereits zielgerichtet angreifen lässt, betrifft das DDR2 Gen. Tumoren mit dieser Mutation können auf eine Therapie mit einem ABL-Inhibitor ansprechen [23]. Dieser TKI wurde ursprünglich als Alternative zu den first line TKI bei der chronischen myeloischen Leukämie entwickelt, zeigt aber durchaus umfassenderes therapeutisches Potenzial. Hier allerdings stößt man diagnostisch auf Probleme. Im Gegensatz zu den Mutationen in EGFR, KRAS oder auch BRAF und PIK3CA, die sich auf wenige Regionen des Gens beschränken, können Mutationen in DDR2 sich über das ganze, sehr große Gen erstrecken. Dies macht es erforderlich, das gesamte Gen zu sequenzieren, um zu einer therapeutischen Entscheidung zu kommen. Hier wird es nötig werden, die neue Technik des Next Generation Sequencing (NGS) in die Diagnostik zu integrieren, die eine Sequenzierung großer Gene erheblich schneller (und kostengünstiger) macht. Das NGS ermöglicht es, dem Patienten und dem behandelnden Arzt die Informationen zum Mutationsstatus in kurzer Zeit zur Verfügung zu stellen, um ein rechtzeitiges Einleiten der Therapie zu ermöglichen. Auch bei den Plattenepithelkarzinomen wurden aber für ungefähr ein Drittel der Tumoren noch keine pathogenetisch relevante Mutation gefunden, sodass die Möglichkeiten einer zielgerichteten Therapie hier noch erheblich eingeschränkter sind als bei den Adenokarzinomen.
Die letzte, bislang unerwähnte Gruppe der kleinzelligen Lungenkarzinome bildet eine vergleichsweise seltene Tumorentität, die stark mit Tabakkonsum assoziiert ist und für die bisher nur wenige potenzielle Targets beschrieben sind. In jüngerer Zeit wurden nun aber auch für diese klinisch schwierige Entität einige genetische Aberrationen beschrieben, die Hoffnung auf neue therapeutische Optionen machen. In 6% der Fälle wurden FGFR1-Amplifikationen beschrieben [13]. Diese haben sich beim Plattenepithelkarzinom (hier in 22% der Fälle) bereits als sensitiv auf FGFR-Inhibition erwiesen [22].
Bei weiteren 10% liegen inaktivierende Mutationen in der Phosphatase PTEN vor. PTEN ist ein negativer Regulator des PI3-Kinase-Signalwegs, dessen Mutationen in der Regel zum Funktionsverlust des Proteins führen. Dies hat zur Folge, dass der PI3-Kinase Signalweg überaktiviert wird, womit sich die Möglichkeit für einen zielgerichteten Therapieansatz eröffnet. Abgesehen von diesen Anfangserfolgen ist der Großteil der pathogenetisch relevanten Mutationen beim kleinzelligen Karzinom jedoch noch unbekannt. Das kleinzellige Lungenkarzinom bleibt daher weiterhin eine klinisch schwierige Gruppe mit äußerst ungünstiger Prognose.
Mit der zunehmenden Kenntnis der genetischen Grundlagen der Lungenkarzinome wird eine immer umfassendere und sensitivere molekulare Diagnostik nötig, die möglichst auch von kleinen Biopsien und zytologischen Präparaten zuverlässige Ergebnisse liefert. Ein wesentliches Hilfsmittel ist hier die Mikrodissektion (manuell oder – noch besser – lasergesteuert), die es ermöglicht, Tumorzellen aus dem umgebenden Normalgewebe zu isolieren und gezielt zu analysieren. Sensitivere Methoden, wie Realtime-PCR-basierte Ansätze und Next Generation Sequencing (NGS), ermöglichen die Detektion von Mutationen auch in Subklonen des Tumors oder bei einem insgesamt geringen Tumoranteil.
Ein weiterer Vorteil des NGS ist es auch, dass viele Gene gleichzeitig und kostengünstig evaluiert werden können. Da viele der neu entdeckten Mutationen nur in einem kleinen Prozentsatz der Patienten gefunden worden sind, ist nur bei gleichzeitiger Testung aller dieser Gene in angemessen kurzer Zeit auch die molekulare Diagnose für den Patienten verfügbar. Neue Techniken, die derzeit entwickelt werden, werden es auch ermöglichen, Punktmutationen, Amplifikationen und Translokationen in einem Ansatz zu untersuchen. Dies lässt hoffen, dass in Zukunft komplexe genetische Analysen des Genoms oder des Exoms helfen werden, auch die bisher unbekannten Mechanismen der Karzinomentstehung und Progression zu ermitteln und ggf. therapeutisch anzugreifen.
Weitere Mutationen bei Adenokarzinomen betreffen z. B. die KRAS-Verwandten HRAS und NRAS, die Rezeptoren HER2 und RET, sowie das Signalmolekül PIK3CA 9,21. Da alle diese Mutationen sehr selten auftreten, sind zielgerichtete Therapien nur für diejenigen von ihnen bekannt, die auch bei anderen Tumoren eine Rolle spielen und bei denen die Therapieoptionen sich auf die Entität der NSCLC übertragen ließen (z. B. PIK3CA-Mutationen und mTOR-Inhibitoren). Andere Mutationen sind bei einzelnen Patienten gefunden worden, aber mehr als ein Drittel der Adenokarzinome können immer noch nicht molekulargenetisch charakterisiert werden. Dies betrifft vor allem die Pathogenese der Rauch-induzierten Karzinome.
Die molekulare Pathogenese der Plattenepithelkarzinome hat sich generell als weniger leicht zugänglich erwiesen, obwohl hier in den letzten Jahren erheblich Fortschritte gemacht wurden. Circa ein Drittel dieser Karzinome zeigen Amplifikationen des PIK3CA-Gens [12]. Hier wird es nötig sein, die molekularen Folgen einer PIK3CA-Amplifikation genauer zu definieren, um zielgerichtet eingreifen zu können.
Eine bessere therapeutische Angriffsfläche bietet die Amplifikation des FGFR1 (fibroblast growth factor receptor 1). Hier wurde bereits ein FGFR-spezifischer TKI entwickelt, der jetzt in klinischen Studien getestet wird [22]. Die hier betroffenen 20-30% der Patienten mit Plattenepithelkarzinomen sollten daher hoffentlich bald von einer zielgerichteten Therapie profitieren können.
Eine Mutation, die spezifisch ist für Plattenepithelkarzinome und die sich heute möglicherweise bereits zielgerichtet angreifen lässt, betrifft das DDR2 Gen. Tumoren mit dieser Mutation können auf eine Therapie mit einem ABL-Inhibitor ansprechen [23]. Dieser TKI wurde ursprünglich als Alternative zu den first line TKI bei der chronischen myeloischen Leukämie entwickelt, zeigt aber durchaus umfassenderes therapeutisches Potenzial. Hier allerdings stößt man diagnostisch auf Probleme. Im Gegensatz zu den Mutationen in EGFR, KRAS oder auch BRAF und PIK3CA, die sich auf wenige Regionen des Gens beschränken, können Mutationen in DDR2 sich über das ganze, sehr große Gen erstrecken. Dies macht es erforderlich, das gesamte Gen zu sequenzieren, um zu einer therapeutischen Entscheidung zu kommen. Hier wird es nötig werden, die neue Technik des Next Generation Sequencing (NGS) in die Diagnostik zu integrieren, die eine Sequenzierung großer Gene erheblich schneller (und kostengünstiger) macht. Das NGS ermöglicht es, dem Patienten und dem behandelnden Arzt die Informationen zum Mutationsstatus in kurzer Zeit zur Verfügung zu stellen, um ein rechtzeitiges Einleiten der Therapie zu ermöglichen. Auch bei den Plattenepithelkarzinomen wurden aber für ungefähr ein Drittel der Tumoren noch keine pathogenetisch relevante Mutation gefunden, sodass die Möglichkeiten einer zielgerichteten Therapie hier noch erheblich eingeschränkter sind als bei den Adenokarzinomen.
Die letzte, bislang unerwähnte Gruppe der kleinzelligen Lungenkarzinome bildet eine vergleichsweise seltene Tumorentität, die stark mit Tabakkonsum assoziiert ist und für die bisher nur wenige potenzielle Targets beschrieben sind. In jüngerer Zeit wurden nun aber auch für diese klinisch schwierige Entität einige genetische Aberrationen beschrieben, die Hoffnung auf neue therapeutische Optionen machen. In 6% der Fälle wurden FGFR1-Amplifikationen beschrieben [13]. Diese haben sich beim Plattenepithelkarzinom (hier in 22% der Fälle) bereits als sensitiv auf FGFR-Inhibition erwiesen [22].
Bei weiteren 10% liegen inaktivierende Mutationen in der Phosphatase PTEN vor. PTEN ist ein negativer Regulator des PI3-Kinase-Signalwegs, dessen Mutationen in der Regel zum Funktionsverlust des Proteins führen. Dies hat zur Folge, dass der PI3-Kinase Signalweg überaktiviert wird, womit sich die Möglichkeit für einen zielgerichteten Therapieansatz eröffnet. Abgesehen von diesen Anfangserfolgen ist der Großteil der pathogenetisch relevanten Mutationen beim kleinzelligen Karzinom jedoch noch unbekannt. Das kleinzellige Lungenkarzinom bleibt daher weiterhin eine klinisch schwierige Gruppe mit äußerst ungünstiger Prognose.
Mit der zunehmenden Kenntnis der genetischen Grundlagen der Lungenkarzinome wird eine immer umfassendere und sensitivere molekulare Diagnostik nötig, die möglichst auch von kleinen Biopsien und zytologischen Präparaten zuverlässige Ergebnisse liefert. Ein wesentliches Hilfsmittel ist hier die Mikrodissektion (manuell oder – noch besser – lasergesteuert), die es ermöglicht, Tumorzellen aus dem umgebenden Normalgewebe zu isolieren und gezielt zu analysieren. Sensitivere Methoden, wie Realtime-PCR-basierte Ansätze und Next Generation Sequencing (NGS), ermöglichen die Detektion von Mutationen auch in Subklonen des Tumors oder bei einem insgesamt geringen Tumoranteil.
Ein weiterer Vorteil des NGS ist es auch, dass viele Gene gleichzeitig und kostengünstig evaluiert werden können. Da viele der neu entdeckten Mutationen nur in einem kleinen Prozentsatz der Patienten gefunden worden sind, ist nur bei gleichzeitiger Testung aller dieser Gene in angemessen kurzer Zeit auch die molekulare Diagnose für den Patienten verfügbar. Neue Techniken, die derzeit entwickelt werden, werden es auch ermöglichen, Punktmutationen, Amplifikationen und Translokationen in einem Ansatz zu untersuchen. Dies lässt hoffen, dass in Zukunft komplexe genetische Analysen des Genoms oder des Exoms helfen werden, auch die bisher unbekannten Mechanismen der Karzinomentstehung und Progression zu ermitteln und ggf. therapeutisch anzugreifen.
Literatur
1. A genomics-based classification of human lung tumors. Sci Transl Med 2013;5(209):209ra153.
2. Mok TS, Wu YL, Thongprasert S et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361(10):947-957.
3. Rosell R, Moran T, Queralt C et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361(10):958-967.
4. Horn L, Pao W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol 2009;27(26):4232-4235.
5. Kwak EL, Bang YJ, Camidge DR et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363(18):1693-1703.
6. Doebele RC, Pilling AB, Aisner DL et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 2012;18(5):1472-1482.
7. Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc 2009;6(2):201-205.
8. Metro G, Chiari R, Baldi A, De A, V, Minotti V, Crino L. Selumetinib: a promising pharmacologic approach for KRAS-mutant advanced non-small-cell lung cancer. Future Oncol 2013;9(2):167-177.
9. Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nat Med 2012;18(3):349-351.
10. Janne PA, Meyerson M. ROS1 rearrangements in lung cancer: a new genomic subset of lung adenocarcinoma. J Clin Oncol 2012;30(8):878-879.
11. Majewski IJ, Mittempergher L, Davidson NM et al. Identification of recurrent FGFR3 fusion genes in lung cancer through kinome-centred RNA sequencing. J Pathol 2013;230(3):270-276.
12. Perez-Moreno P, Brambilla E, Thomas R, Soria JC. Squamous cell carcinoma of the lung: molecular subtypes and therapeutic opportunities. Clin Cancer Res 2012;18(9):2443-2451
13. Peifer M, Fernandez-Cuesta L, Sos ML et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012;44(10):1104-1110.
14. Samowitz WS, Sweeney C, Herrick J et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res 2005;65(14):6063-6069.
15. Marchetti A, Felicioni L, Malatesta S et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol 2011;29(26):3574-3579.
16. Flaherty KT, Puzanov I, Kim KB et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363(9):809-819.
17. Prahallad A, Sun C, Huang S et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483(7387):100-103.
18. Yu HA, Arcila ME, Rekhtman N et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19(8):2240-2247.
19. Cappuzzo F, Marchetti A, Skokan M et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol 2009;27(10):1667-1674.
20. Kong-Beltran M, Seshagiri S, Zha J et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res 2006;66(1):283-289.
21. Kohno T, Ichikawa H, Totoki Y et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med 2012;18(3):375-377.
22. Reck M, Kaiser R, Mellemgaard A et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol 2014;15(2):143-155.
23. Hammerman PS, Sos ML, Ramos AH et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov 2011;1(1):78-89.

Dr. med. Markus Tiemann
Institut für Hämatopathologie Hamburg
Fangdieckstraße 75a, 22547 Hamburg
Tel.: 040/707085-300
Fax: 040/707085-210
mtiemann[at]hp-hamburg[dot]de