Das Multiple Myelom ist eine maligne hämatologische Erkrankung, verursacht durch die Proliferation entarteter Plasmazellen. Die Diagnostik umfasst neben Anamnese und körperlicher Untersuchung vor allem Blut- und Urintests sowie Knochenmarkausstriche, welche auch zur Stadieneinteilung und Verlaufskontrolle dienen. Die Entscheidung für eine systemische Therapie erfolgt in der Regel erst, wenn Organschäden eingetreten sind oder kurz bevorstehen. Die Therapie wird individuell u. a. anhand von biologischem Alter, Ko-Morbiditäten und Laborbefunden des Patienten festgelegt.
Schlüsselwörter: Multiples Myelom, M-Gradient, Immunfixation, freie Leichtketten, Plasmazellen
Das Multiple Myelom (MM) ist eine derzeit noch meist unheilbare, maligne hämatologische Systemerkrankung, die in der Systematik der Weltgesundheitsorganisation (WHO) zu den lymphoproliferativen B-Zellerkrankungen gerechnet wird [1]. Ihre Inzidenz liegt weltweit bei etwa 6–7 Fällen pro 100.000 Einwohnern pro Jahr; in Deutschland zählt das Multiple Myelom damit zu den 20 häufigsten Tumorerkrankungen. Pathophysiologische Grundlage ist die unkontrollierte Vermehrung monoklonaler Plasmazellen, weshalb die Erkrankung auch als Plasmozytom bezeichnet wird. Die Proliferation ist i. d. R. auf das Knochenmark beschränkt und führt zu einer Produktion von intakten, aber funktionslosen Immunglobulinen und/oder freien leichten Ketten eines Immunglobulins [2].
Pathogenese und Klinik
Das Multiple Myelom (MM) wird zwar als eine Krankheitsentität gesehen, ist aber genetisch und klinisch sehr heterogen. Die wichtigsten Untergruppen sind die MGUS (Monoklonale Gammopathie Unklarer Signifikanz), das „schwelende Myelom“ (Smoldering Myeloma, SMM) und das klinisch manifeste MM (Tab. 1). Bei MGUS kann im Blut ein Paraprotein, also ein monoklonaler Antikörper nachgewiesen werden; die Patienten zeigen jedoch keine Krankheitszeichen. Das Risiko für den Übergang von einer MGUS in ein Multiples Myelom liegt bei 1 bis 1,5% pro Jahr.
Beim Smoldering Myelom ist zwar der Anteil pathologischer Plasmazellen im Knochenmark deutlich erhöht oder ein Paraprotein stark vermehrt, jedoch ohne klinische Symptome oder Zeichen einer Organbeteiligung.
Die Auslöser der Entartung der Plasmazellen sind weitgehend unbekannt, doch bei immerhin etwa 40% der Patienten findet man zytogenetisch Trisomien verschiedener Chromosomen. Translokationen mit Beteiligung des Immunglobulin-Schwerkettengens (IgH) treten bei vielen nicht-hyperdiploiden Myelomen im Chromosom 14q32 auf. Häufig sind auch reziproke Translokationen wie z. B. t(11;14), t(4;14) oder t(14;16). Diese primären genetischen Aberrationen können auch bei Patienten mit MGUS nachgewiesen werden. Die genetischen Aberrationen haben Einfluss auf das klinische Bild, die Prognose und das Ansprechen auf verschiedene Therapien.
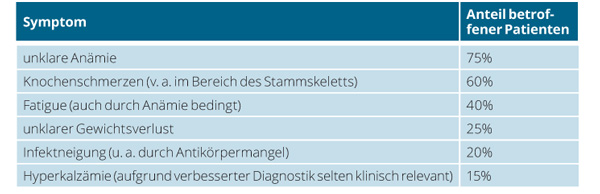
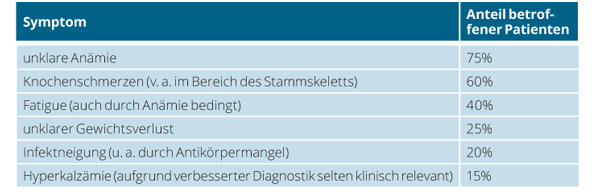
Patienten mit Multiplem Myelom suchen den Arzt häufig wegen unspezifischer Symptome, die meist schon über einen längeren Zeitraum bestehen, auf (Tab. 2). Die Diagnostik des Multiplen Myeloms ist eine Domäne der Labormedizin, umfasst aber neben der wegweisenden Anamnese und körperlichen Untersuchung auch die Bildgebung. Diese dient vor allem zur Erfassung von Knochenveränderungen. Hier wurde das konventionelle Ganzkörperröntgen nach „Pariser Schema“ weitgehend durch die „low dose“-Ganzkörper-Computertomografie oder die deutlich sensitivere Ganzkörper-Magnetresonanztomografie abgelöst.