Domäne der Immunhistochemie
Hodenkrebs
Testikuläre Keimzelltumoren weisen eine vielfältige Morphologie auf. Bei exakter Diagnose und verlässlichem Staging bestehen heute jedoch exzellente Heilungs- und Überlebenschancen.
Hodenkrebs gehört mit gut 1% aller männlichen Tumoren und einer jährlichen Neuerkrankungsrate von weniger als zehn pro 100.000 Männer in den westlichen Ländern zur Kategorie der seltenen Erkrankungen. 95% davon sind Keimzelltumoren (KZT), die also von Zellen der Spermiogenese abstammen; der Rest entfällt auf Keimstrang-Stroma-Tumoren oder tumoröse Veränderungen anderen Ursprungs, zum Beispiel Lymphome oder Metastasen.
Im Gegensatz zu fast allen anderen Krebserkrankungen tritt Hodenkrebs in relativ jungen Jahren, nämlich zwischen 25 und 45, auf. In dieser Altersgruppe ist er nach Angaben des Robert-Koch-Institus der häufigste bösartige Tumor bei Männern in Deutschland. Weltweite Übersichtskarten zeigen ein ausgeprägtes West-Ost- und Nord-Süd-Gefälle. In Mittel- und Nordeuropa findet man die höchsten Erkrankungsraten überhaupt; die niedrigsten Raten haben afrikanische Eingeborene. Weiße erkranken insgesamt häufiger als Asiaten, und diese häufiger als Schwarzafrikaner.
Und noch eines ist erstaunlich: Noch vor 30 Jahren kam die Diagnose Hodenkrebs einem Todesurteil gleich. Heute weisen KZT trotz steigender Inzidenzen die niedrigste Mortalität unter allen malignen Tumoren auf. Die 5-Jahres-Überlebensrate liegt in Europa bei 96% (alle Stadien eingerechnet). Zu dieser positiven Entwicklung trägt zum einen ein sorgfältiges Staging zum Zeitpunkt der Diagnose bei, zum anderen die Möglichkeiten einer stadiengerechten Therapie mit verschiedenen Kombinationen aus Operation, Chemo- und Strahlentherapie, ein engmaschiges Follow-up und gegebenenfalls aggressive Salvage-Therapien.
Die statistisch wohl am besten gesicherten Risikofaktoren sind Kryptorchismus, gonadale Dysgenesie, Klinefelter Syndrom sowie KZT bei Verwandten ersten Grades (wobei das Risiko für Männer mit erkrankten Brüdern höher ist als das für Männer mit erkranktem Vater). Für hormonelle Einflüsse gibt es Indizien, zu den Einflüssen von Ernährung und Beruf liegen keine verlässlichen Daten vor.
Genetische Veränderungen
Aktuelle Theorien besagen, dass atypische Gonozyten bereits intrauterin entstehen. Sie behalten im Gegensatz zu gesunden Keimzellen ihre Stammzelleigenschaften und somit ihr pluripotentes Entwicklungspotenzial bei, bis sie sich durch weitere, nach der Geburt einwirkende Noxen zunächst zu intratubulären, dann zu invasiven KZT weiterentwickeln.
Hervorzuheben ist eine typische genetische Veränderung, die sowohl in der intratubulären Keimzellneoplasie (IGCNU) als auch in allen histologischen Typen invasiver Keimzelltumoren beobachtet wird, nämlich die Bildung eines sogenannten Isochromosoms i(12p) mit Zugewinnen am kurzen Arm von Chromosom 12. Dieses enthält Gene, die den malignen Zellen Proliferations- und Überlebensvorteile verleihen, zum Beispiel das Onkogen K-RAS oder den Transkriptionsfaktor SOX5.
Seminome
Seminome sind mit etwa 50% die häufigsten KZT des Hodens. Unterschieden werden das klassische Seminom und das sehr seltene spermatozytische Seminom. Die Herausforderung für den Pathologen liegt in einer exakten Abgrenzung gegenüber nicht-seminomatösen KZT (s. u.) bzw. dem Erkennen einer nicht-seminomatösen Tumorkomponente in einem ansonsten klassischen Seminom, was für Prognose und Therapie entscheidend ist.
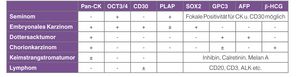
Die typische Morphologie ist hier beschrieben. Da durchaus Abweichungen vom klassischen Bild vorkommen, stellt die Immunhistochemie ein oft unentbehrliches differenzialdiagnostisches Werkzeug dar. In den S1-Leitlinien[1] wird ein Markerpanel von minimal drei Antikörpern empfohlen: Panzytokeratin (Identifikation nicht-seminomatöser Tumoranteile), OCT3/4 oder D2-40 (Erkennung einer IGCNU) und CD31 (Identifikation einer Angioinvasion). Weitere Marker können fakultativ zur genaueren Abgrenzung einzelner Tumorkomponenten verwendet werden (siehe Tabelle).
Zumeist befinden sich testikuläre KZT bei Erstdiagnose im Stadium I, sind also auf Hoden und eventuell Nebenhoden begrenzt und weisen keine Angioinvasion oder Metastasen auf. Für Seminome im Stadium I ist die Prognose exzellent, sodass in vielen Fällen die operative Therapie in Form einer radikalen Orchiektomie ausreichend ist.
In der Literatur wird beschrieben, dass Seminome mit einer Größe von über 4 cm oder mit Infiltration des Rete testis ein erhöhtes Rezidivrisiko zeigen. Obwohl prospektive Studien hierzu noch ausstehen, sollten beide Parameter im pathologischen Befund erwähnt werden. Interessanterweise wird der Nachweis einer Angioinvasion bei der Risikostratifizierung von Seminomen (im Gegensatz zu nicht-seminomatösen KZT) eher zurückhaltend diskutiert.
Nicht-seminomatöse KZT
Die übrigen 50% der testikulären Keimzelltumoren verteilen sich auf vier Tumorentitäten, die im Erwachsenenalter nur sehr selten in Reinform auftreten, sondern meist als Komponenten eines gemischten KZT vorkommen. Das typische Zellbild des embryonalen Karzinoms ist auf S. 219 dargestellt. Hier liegt zum Zeitpunkt der Erstdiagnose häufig bereits eine Lymph- oder Hämangioinvasion vor.
Der Dottersacktumor in Reinform ist der nach dem Teratom zweithäufigste KZT des Kindesalters und ausnahmslos maligne. Beim Erwachsenen kommt er in 40% der gemischten KZT als Teilkomponente vor. In der Regel geht er mit einer deutlichen Erhöhung von AFP im Serum einher. Für den Pathologen stellt diese Form eine besondere Herausforderung dar, denn man kann innerhalb ein und desselben Tumors ein Nebeneinander unterschiedlicher Wachstumsmuster beobachten, die häufig erst immunhistochemisch derselben Tumorentität zugeordnet werden können.
Eine besondere Entität innerhalb der nicht-seminomatösen KZT stellt das Teratom dar, das aus Gewebsabkömmlingen aller drei Keimblätter, also aus Ekto-, Ento- und Mesoderm, bestehen kann. Es ist in Reinform der häufigste KZT im Kindesalter und als benigne anzusehen. Dagegen sind im Erwachsenenalter alle „echten“ Teratome von einer IGCNU begleitet und – im Gegensatz zu Teratomen des Ovars – stets maligne.
Teratome kommen in etwa 50% der gemischten KZT vor; selten enthalten diese eine maligne Gewebskomponente, die man normalerweise von extratestikulären Geweben kennt, z. B. in Form eines Plattenepithelkarzinoms. Man spricht von einem Teratom mit „somatischer Malignität“, wenn bei schwacher Vergrößerung (4er-Objektiv) in mehr als der Hälfte eines Gesichtsfeldes maligne Tumorformationen nachgewiesen werden können.
Diese Komponente bestimmt bei Auftreten von Metastasen entscheidend die Prognose, sodass ein Erkennen und die eindeutige Zuordnung wichtig sind. Ist eine Abgrenzung gegenüber Metastasen eines anderweitig lokalisierten Primarius schwierig, dann kann die Bestimmung des Isochromosoms i(12p) die Herkunft aus einem KZT beweisen.
Das seltene Chorionkarzinom besteht aus zwei Komponenten, den intermediären Trophoblast- und Zytotrophoblastzellen und aus synzytiotrophoblastären Riesenzellen. Da letztere manchmal auch in anderen KZT vorkommen, ist der Nachweis beider Zelltypen für die Diagnose entscheidend. Chorionkarzinome können ausgedehnte Einblutungen und Nekrosen aufweisen, die von einer anderweitig verursachten Gewebsschädigung – z. B. durch Trauma – abgegrenzt werden müssen. Die Prognose ist vor allem im metastasierten Stadium deutlich schlechter als die anderer KZT und korreliert mit der Höhe des Serum-β-HCG-Spiegels, der die Tumorlast widerspiegelt.
Besteht ein KZT aus mehr als einem der genannten histologischen Typen, liegt per definitionem ein gemischter KZT vor. Dies ist in knapp 70% der nicht-seminomatösen und in ca. 30% aller KZT der Fall. Im pathologischen Befundbericht sollte man die verschiedenen Tumorkomponenten auf-listen und den jeweiligen Prozentanteil am Gesamttumor angeben.
Als ungünstige Prognosefaktoren für nicht-seminomatöse KZT werden das Vorliegen einer Angioinvasion, eine Proliferationsrate von mehr als 70% in der Ki67-Färbung und ein Anteil von embryonalem Karzinom über 50% angegeben.