Das NETz wird enger
Hämostase und Inflammation
Gerinnungs- und Entzündungsvorgänge greifen bei der Entstehung und Heilung vieler Krankheiten ineinander. Jetzt wird dieses komplizierte Netzwerk durch die Entdeckung der neutrophile extracellular traps (NETs) noch enger geknüpft.
Dass die Gerinnungskaskade in natura ebenso wie bei Labortests im Blutplasma abläuft und ausschließlich für die Blutstillung zuständig ist, entspricht nicht mehr den heutigen Vorstellungen. Vielmehr wirken Gerinnungsfaktoren und andere Plasmaproteine sowie Zellen des Blutes und der Gefäßwand eng zusammen, um nicht nur den oft zitierten „Schnitt in den Finger“ mit einem Gerinnsel zu verschließen, sondern auch zerstörte Zellen abzubauen, Fremdstoffe zu neutralisieren, Erreger abzuwehren und Gewebe zu reparieren.
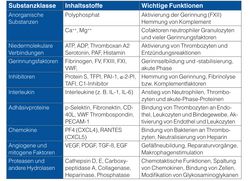
Diese intensive Interaktion verschiedener Signal- und Stoffwechselwege lässt sich sehr gut am Beispiel der Thrombozyten demonstrieren (siehe Tabelle): Sie initiieren primär den Wundverschluss durch einen Thrombus, sind aber auch in zahlreiche Entzündungs- und Regenerationsvorgänge involviert.
Das Zusammenspiel zwischen Hämostase und Inflammation spiegelt sich in vielen labormedizinisch relevanten Konstellationen wider: So können erhöhte Fibrinogenspiegel für eine Thromboseneigung im Rahmen chronischer Entzündungen sprechen, denn Fibrinogen ist auch ein Akute-Phase-Protein. Bei Thromboembolien sieht man häufig einen gleichzeitigen Anstieg von CRP und D-Dimeren als Zeichen der entzündlichen Reaktion und gesteigerten Fibrinolyse. D-Dimere können dabei sowohl von Plasmin im Rahmen der klassischen Fibrinolyse, als auch von PMN-Elastase im Rahmen einer entzündlichen Proteolyse gebildet werden.
Aktivierung der Hämostase
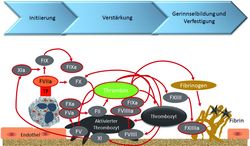
Wo – vom Schnitt in den Finger einmal abgesehen – solche komplexen Prozesse ihren Ausgang nehmen, ist gar nicht so leicht einzugrenzen. Einige aktivierte Gerinnungsfaktoren sind Initiatoren der Entzündung – und umgekehrt. Exogene und endogene Trigger wie etwa Bakterien oder atherosklerotische Plaques führen unter anderem zur Ausschüttung der Zytokine TNF-α und IL-1-β, die Endothelzellen und Monozyten aktivieren und die Hämostase induzieren: Adhäsivproteine fördern die Bindung von Thrombin an die geschädigte Gefäßwand, Tissue Factor (TF) verstärkt die Entzündung und aktiviert die Gerinnungsfaktoren FVII und FIX, sodass Prothrombin über das extrinsische und intrinsische System zu Thrombin gespalten wird (siehe Abbildung).
TFPI (Tissue Factor Pathway Inhibitor) unterdrückt auf dieser Stufe eine unkontrollierte Aktivierung der Gerinnung. Erst durch die Beteiligung von Thrombozyten wird sie so verstärkt, dass am Ende tatsächlich Thrombin entsteht. Dieses kann dann seine eigene Bildung über die Aktivierung weiterer Gerinnungsfaktoren im Sinne einer positiven Rückkopplung massiv steigern. Entscheidend daran beteiligt ist Faktor VIII, der wie auch CRP oder Fibrinogen zu den Akute-Phase-Proteinen gehört.
Regulation der Hämostase
Die Bildung von Thrombin wird durch Protease-Inhibitoren wie TFPI, Antithrombin (AT) oder Protein C gegenreguliert. AT neutralisiert aktivierte Gerinnungsfaktoren und verfügt zudem über direkte antiinflammatorische Eigenschaften. Wesentlich komplizierter ist der Wirkungsmechanismus von aktiviertem Protein C(APC). Es spaltet die „Motoren“ der Gerinnungskaskade, FVIIIa und FVa, die für eine mehr als tausendfache Beschleunigung der jeweiligen Reaktionen sorgen. Diese Spaltung läuft auf der Oberfläche von Endothel unter Beteiligung des Protein C-Rezeptors (EPCR) ab und erfordert freies Protein S (PS). PS wiederum kann in der akuten Phase durch verstärkte Komplexierung am C4b-Binding Protein (Komplementsystem) seine Wirkung verlieren, sodass die Entzündung indirekt die Thrombinbildung begünstigt. Proteine, die für die Hemmung der Hämostase sorgen (z. B. AT, APC), werden daher bei Entzündungen verbraucht, was die Bildung von Thrombosen fördern kann.
Fibrinbildung und Fibrinolyse
Thrombin spaltet nicht nur Fibrinogen zu Fibrin, sondern aktiviert auch TAFI (Thrombin-activatable Fibrinolysis Inhibitor) und FXIII. Diese beiden Proteine stabilisieren das Gerinnsel und FXIII fixiert es zusätzlich an Bindegewebe und anderen Oberflächen. Ähnlich stabilisierend wirken auch zwei Komponenten des Entzündungssystems, nämlich die Zytokine IL-1 und TNF-α, indem sie an der Gefäßwand in die Regulation der Fibrinolyse eingreifen: Der Fibrinolyseaktivator t-PA (Tissue Plasminogen Activator) wird herunterreguliert, während die Biosynthese seines Gegenspielers PAI-1 (Plasminogen Activator Inhibitor) erheblich zunimmt. Somit hemmen Entzündungen die Fibrinolyse und beeinflussen indirekt viele weitere Vorgänge, die durch fibrinolytische Enzyme ausgelöst werden. Davon betroffen sind unter anderem die Angiogenese, Apoptose und Wundheilung.
Die in den letzten beiden Abschnitten beschriebenen Zusammenhänge haben unter anderem bei der Sepsis große pathophysiologische und diagnostische Bedeutung. Hier kann die Interaktion zwischen Gerinnungs- und Entzündungsvorgängen zu disseminierten Mikrogerinnseln mit massivem Verbrauch von Gerinnungsfaktoren und Inhibitoren sowie zu Organversagen führen. Für die Erkennung dieses kritischen Zustands haben sich neben Gerinnungstests und D-Dimer auch PAI-1 sowie insbesondere die Kombination von Protein C und Presepsin als Biomarker bewährt.
Die Rolle der Thrombozyten
Thrombin, bakterielle Polysaccharide, Histone aus zerfallenden Zellen und andere Substanzen können Thrombozyten aktivieren und damit die Gerinnung und Entzündung fördern. Dabei werden aber gleichzeitig auch verschiedenste Inhalte aus dem Zytosol oder speziellen Granula freigesetzt (siehe Tabelle). Nur ein Bruchteil davon hat direkt mit Gerinnung zu tun, aber viele spielen eine Rolle in der Inflammation und Immunabwehr.
Ein wichtiges, auch diagnostisch verwertbares Signal für die Aktivierung von Thrombozyten ist die verstärkte Expression von p-Selektin (siehe Abbildung). Dieser Rezeptor kann über seinen Gegenspieler CD-40L Monozyten binden, aktivieren und so durch Expression von TF die Gerinnung weiter ankurbeln. Während einer Sepsis kommt es daher oft zu einer ausgeprägten Thrombozytopenie; mittels Flowzytometrie findet man Komplexe aus Thrombozyten und Monozyten, die sich diagnostisch verwenden lassen.
Von pathophysiologischer Bedeutung sind schließlich auch Mikropartikel, die aus aktivierten Thrombozyten und Monozyten freigesetzt werden. Insbesondere wenn sie Tissue Factor tragen, können sie die Gerinnungs- und Entzündungsaktivierung mit dem Blutstrom weitertragen und an der Entwicklung einer disseminierten intravaskulären Gerinnung beteiligt sein.
Neutrophile Netze
Gerinnsel dienen nicht nur dem Wundverschluss, sondern können auch eingedrungene Erreger einschließen, um eine ungehemmte Ausbreitung zu verhindern. Im Gegenzug haben einige Bakterien Enzyme wie zum Beispiel Streptokinase entwickelt, die Gerinnsel auflösen können – eine Erfindung der Natur, die man sich bei thrombembolischen Ereignissen auch therapeutisch zunutze macht.
Noch bekannter ist das breite Arsenal von Abwehrmechanismen der Leukozyten bei der Infektionsbekämpfung. Hier entdeckten Max-Planck-Forscher um Dr. Volker Brinkmann, Berlin, gemeinsam mit New Yorker Kollegen 2004 ein überraschendes Phänomen: Neutrophile Granulozyten zerstören eingedrungene Pathogene im Rahmen der innaten (angeborenen) Immunabwehr nicht nur durch Phagozytose unter Beteiligung des Komplements, sondern werfen auch „Fangnetze“ aus, die sich von Fibrinnetzen biochemisch grundsätzlich unterscheiden. Ihre Hauptbestandteile sind bis zu 50 nm lange DNA-Fäden, in deren Maschen sich ein giftiger Proteincocktail aus Histonen, Enzymen und Peptiden befindet.
Eingeleitet wird dieser als NETose bezeichnete Abwehrmechanismus unter anderem durch Thrombozyten. Sie binden über den TLR4-Rezeptor und andere Mediatoren an Bakterien, Pilze, Viren und auch Parasiten und aktivieren die Neutrophilen unter Mitwirkung von IL-8. Die Berliner Forscher beobachteten im Elektronenmikroskop eine Abflachung der Leukozyten, gefolgt von einer Dekondensierung des Chromatins. Anschließend lösen sich Kern- und Zellmembran unter maßgeblicher Mitwirkung von freien Radikalen (ROS = Reactive Oxygen Species) auf und entlassen die sogenannten Neurophil Extracellular Traps („neutrophile extrazelluläre Fallen“, NETs). Die Zellen selbst gehen dabei zugrunde. Doch dieses Opfer lohnt sich für den Gesamtorganismus: Ihr aus dem Kern und Zytosol sowie den Granula stammender Inhalt ist in der Lage, die Eindringlinge abzutöten und aufzulösen.
Pathophysiologie der NETs
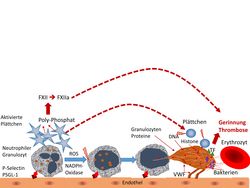
Bevölkerungsstudien bestätigen, dass nach Infektionen gehäuft Thrombosen auftreten. Daran sind offenbar Thrombozyten, Neutrophile und auch NETs ursächlich beteiligt. Auf aktivierten Endothelzellen binden Letztere an hochmultimere, besonders thrombogene Moleküle des von Willebrand Faktors, an die sich wiederum Thrombozyten und weitere Neutrophile anlagern. Durch TF in Monozyten kann die Aktivierung der Gerinnung und weiterer Thrombozyten ausgelöst werden, und dieser Vorgang wird durch freigesetzte DNA und Polyphosphate aus Thrombozytengranula verstärkt, die FXII aktivieren (Abbildung oben). Das ist zwar für die Blutstillung nicht erforderlich, erhöht aber die Stabilität des Thrombus.
NET-Material findet sich in Thromben, und Sepsispatienten weisen auch im Plasma erhöhte Spiegel von Biomarkern wie etwa MPO (Myeloperoxidase), Elastase, Histone oder zellfreie DNA auf, die aus den NETs stammen. Heparin reduziert – vermutlich durch seine stark negative Ladung – die Anheftung von Plättchen an die NETs und bindet selbst an Histone, was wohl einen Teil der antiinflammatorischen Wirkungen von Heparin erklärt.
So wichtig NETs für die innate Immunabwehr sind, so haben sie doch auch ihre Schattenseiten. Patienten mit systemischem Lupus erythematodes (SLE) bilden bei bestimmten Infektionen – beispielsweise einer Malaria – Autoantikörper gegen DNA und Histone sowie Proteine aus den Granula von Neutrophilen aus. Eine neue Infektion mit NET-Bildung kann dann einen SLE-Schub auslösen. NETs sind wohl auch an der Entstehung der Prä-Eklampsie beteiligt; man vermutet, dass bei einer Entzündung in der Schwangerschaft NETs im Bereich der Kontaktfläche zwischen Mutter und Embryo entstehen, die die Blut- und Sauerstoffversorgung des Embryos gefährden.
Umgekehrt hat auch ein Defekt in der NET-Bildung krankhafte Folgen: Bei der chronischen Granulomatose ist die NETose aufgrund eines Mangels an NADPH-Oxidase gestört. In der Folge kommt es bei solchen Patienten häufiger zu Infektionen. Die Transfusion-Related Acute Lung Injury (TRALI) ist eine seltene, aber gefährliche Nebenwirkung von Bluttransfusionen, bei der neben Auto-Antikörpern NETs eine entscheidende Rolle spielen.
Künftige Therapieoptionen
Die Reduktion von NETs wird derzeit als Therapieoption bei Thrombosen und thrombotischen Mikroangiopathien diskutiert. Als präventive Maßnahme kommt eine pharmakologische Hemmung der Enzyme Peptidylarginin-Deiminase (PAD-4, verantwortlich für die Citrullinierung von Histonen) oder NADPH-Oxidase (Produktion von ROS) infrage, um die Bildung von NETs zu vermindern. Zum Abbau bereits existierender NETs testet man eine rekombinante Protease rADAMTS 13; ein angeborener oder erworbener Mangel an ADAMTS 13 wurde bei der thrombotisch-thrombozytopenischen Purpura bzw. bei Herzinfarkt und Sepsis beobachtet.
Schließlich wehren sich auch einige Bakterienarten mithilfe von Katalase oder DNAsen gegen NETs – möglicherweise eine weitere Erfindung der Natur, die man sich beim Kampf gegen Atherosklerose, Thrombosen und andere Folgen akuter und chronischer Entzündungen zunutze machen könnte.

Dr. Hans-Jürgen Kolde
Mitglied des Fachbeirats