Disruptive Innovationen
Die Zukunft der hämato-onkologischen Diagnostik
Weiterhin bleibt die phänotypische Beschreibung hämatologischer Neoplasien der Goldstandard der Diagnostik und Therapiekontrolle, doch molekulardiagnostische Verfahren spielen eine immer größere Rolle. Die zu erwartende Datenflut wird das Fach in den nächsten Jahren grundlegend verändern und der Bioinformatik eine Schlüsselrolle zuweisen.
Schlüsselwörter: Molekulardiagnostik, Bioinformatik, künstliche Intelligenz
Seit November 2017 liegt die neue WHO-Klassifikation der hämatologischen Neoplasien und Lymphome vor [1]. Mit knapp 600 Seiten und 4.500 Literaturangaben stellt sie das aktuelle Standardwerk für die Diagnostik, Klassifikation und vielfach auch prognostische Einschätzung dar. Wie schon seit mehr als 150 Jahren basiert die Einteilung primär auf dem Phänotypen der Zellen im Blut, Knochenmark oder den Lymphknoten, doch zunehmend gewinnt nun auch die Molekulardiagnostik an Bedeutung.
Der Blick ins Mikroskop bleibt unverändert und bis auf Weiteres der Goldstandard zur Diagnose hämatologischer Neoplasien. Doch in den letzten 50 Jahren wurden die rein panoptischen Färbungen (z. B. nach Pappenheim oder im HE-Schnitt) durch die Einführung zytochemischer Reaktionen (Myeloperoxidase, Chloracetat-Esterase, Eisenfärbung etc.) immer spezifischer. Diese Verfahren sind zeitnah zu erbringen, im Vergleich zu vielen neuen Methoden kostengünstig und leicht lokal vorzuhalten. Andererseits bedürfen sie hoher Expertise und Erfahrung: Gerade das Antrainieren phänotypischer Engramme und deren Bezug zu anderen Methoden kostet sehr viel Zeit, und seitens der angehenden Hämatologen und Pathologen erfordern sie eine gewisse Begabung.
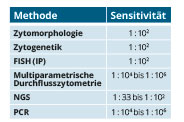
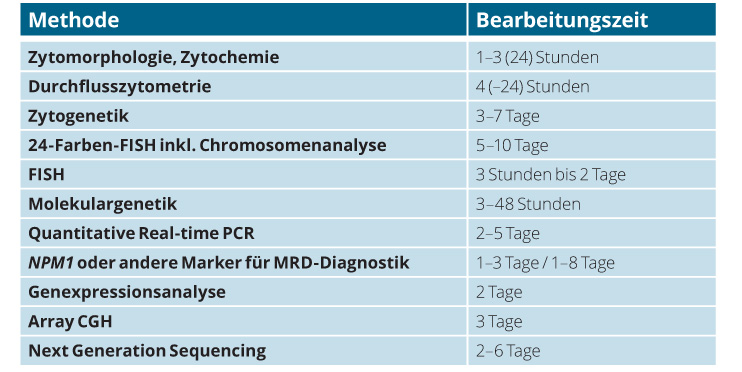
Gleichzeitig muss analytisches Wissen, etwa zu den Nachweisempfindlichkeiten der Chromosomenanalyse, Fluoreszenz-in-situ-Hybridisierung (FISH), Immunhistologie und Immunphänotypisierung erworben werden (Tab. 1), und die rasch steigende Zahl neuer molekularmethodischer Verfahren macht es nicht einfach, immer auf dem neuesten Stand zu bleiben [2]. Dessen ungeachtet bleibt im Hinblick auf alle weiteren diagnostischen Strategien die phänotypische Beschreibung der malignen Zellen richtungsweisend.
Die molekulare Ebene
Immunhistochemie und Immunphänotypisierung erschlossen der Diagnostik in den letzten 40 Jahren die molekulare Ebene der Proteine und machten die Aussagen sehr viel granulierter, sodass man heute in vielen Fällen allein durch Kombination mit der panoptischen Färbung Leukämien und Lymphome ausreichend sicher diagnostizieren kann.
Für die neue Klassifikation nach WHO und insbesondere für die Definition von Prognosekriterien reicht dies aber nicht mehr aus. Immer stärker muss nun die genomische Ebene einbezogen werden; insbesondere in der Therapie nimmt unter dem Schlagwort Precision Medicine die Zahl der Medikamente rasch zu, deren Zulassung mit dem Nachweis eines Zielmoleküls verknüpft ist. Hier sind in der Regel molekulardiagnostische Tests (Companion Diagnostics) durchzuführen, ehe der behandelnde Arzt das Rezept unterschreibt.
Ein weiteres wichtiges Feld der molekular getriebenen Diagnostik ist die minimale Resterkrankung (MRD = Minimal Residual Disease, neuerdings Measurable Residual Disease). Diese kann mit molekulargenetischen Methoden bestimmt werden oder auch mit immunphänotypischen Analysen, z. B. beim sog. Leukämie-assoziierten Immunphänotyp (LAIP).
Zytogenetik
Auch die Chromosomenanalyse auf Basis klassischer Metaphasen erfordert hohe Sachkenntnis. Pro Fall sind bis zu fünf viable Zellkulturen parallel anzulegen, die nach ein bis drei Tagen in der Metaphase blockiert werden und erst durch weitere Analyseschritte zum Legen des Karyogramms zur Verfügung stehen [3]. Die organisatorische Anforderung unterscheidet sich von allen anderen Ansätzen, weil die Proben rasch ins Labor gebracht werden müssen, um lebende Leukämie- oder Lymphomzellen in Zellteilung bringen zu können.
Die dabei erhaltenen Ergebnisse sind heute zentraler Bestandteil der Diagnostik vieler Leukämien und auch Lymphome. Das erste und nach wie vor prominenteste Beispiel war der Nachweis des sogenannten Philadelphia Chromosoms [4], später als t(9;22) erkannt und noch später mit dem molekularen Korrelat BCR-ABL1 als Fusionsgen genauer beschrieben. Inzwischen gehört die klassische Chromosomenanalyse zur Routinediagnostik – speziell bei den akuten Leukämien (AML, ALL), aber natürlich auch bei der CML und zunehmend bei der CLL. Sie unterstützt die aufgrund des Phänotyps am Mikroskop oder mit der Immunphänotypisierung gestellte Diagnose und ist für die Klassifikation nach WHO ebenso wichtig wie für die verwendeten Prognose-Scores, zum Beispiel ELN für die AML oder IPSS-R für MDS [5, 6].
In den letzten 30 Jahren kam zur klassischen Chromosomenanalyse die molekulare Zytogenetik (FISH) als Ergänzung oder auch Alternative hinzu. Ihr größter Vorteil: Sie kann an Interphase-Zellkernen durchgeführt werden, sodass die Zellkultur zur Generierung von Metaphasen nicht mehr in allen Fällen erforderlich ist. Der Zeitbedarf sinkt dadurch von drei bis sieben auf ein bis zwei Tage.
Die Sensitivität des FISH-Verfahrens ist etwas höher als die der klassischen Chromosomenanalyse. Dafür kann man hier allerdings nur das detektieren, was man vorher auch gefragt hat, da man typischerweise mit einzelnen FISH-Sonden ganz bestimmte Gene oder Chromosomenabschnitte wie etwa BCR-ABL1, Trisomie 8 oder PML-RARA adressiert.
Molekulargenetik
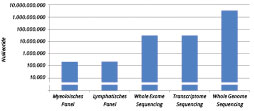
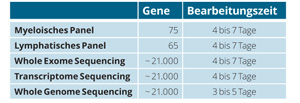
Inzwischen hat sich die Molekulargenetik fest in das Feld der diagnostischen Möglichkeiten eingereiht – beginnend mit dem Nachweis von Fusionsgenen und einzelnen Basenaustauschen über Copy Number Variations (CNV) bis hin zum Whole Exome Sequencing (WES), Whole Genome Sequencing (WGS) und der Sequenzierung der kompletten RNA (RNA-Seq). Aus Abb. 1 und Tab. 2 wird die Spannweite der erfassten Genombereiche über mehrere Zehnerpotenzen hinweg ersichtlich. Die erreichbare Sensitivität liegt bei 1 : 104 bis 1 : 106 und somit in derselben Größenordnung wie die ähnlich sensitive Immunphänotypisierung.
Aus der neuen WHO-Klassifikation wird klar, dass gegenwärtig ein diagnostischer Paradigmenwechsel vom Phänotyp zum Genotyp stattfindet. Molekulargenetische Befunde werden künftig bei vielen hämatologischen Neoplasien die entscheidende Rolle für Diagnose und Prognose sowie Therapie und Therapiekontrolle spielen. Ein prominentes Beispiel ist die V617F-Mutation im JAK2-Gen, die zu einer dysregulierten Aktivierung des JAK-STAT-Signalwegs führt. Ein und derselbe molekulare Mechanismus zieht hier phänotypisch ganz unterschiedliche myeloproliferative Erkrankungen wie etwa Polycythämia vera, essenzielle Thrombozythämie oder primäre Myelofibrose nach sich. Umgekehrt kommt es aber auch bei Patienten, die keine JAK2-Mutation aufweisen, zur Aktivierung des JAK-STAT-Signalwegs – mit entsprechenden Konsequenzen für die gezielte Therapie.
Wohin geht die Entwicklung?
Die wichtige Frage, die sich derzeit stellt, ist vor allem: Wie kann man phänotypische und genotypische Methoden im Interesse der Patienten am besten kombinieren? Wohin könnte die Entwicklung gehen? Dabei geht es keineswegs nur um einen qualitativen Paradigmenwechsel. Vielmehr nimmt auch die Zahl der notwendigen Analysen rasch zu, nicht nur bei der Erstdiagnose, sondern auch im Rezidiv und im Verlauf – hier speziell zur Bestimmung der minimalen Resterkrankung.
Das bedeutet, dass man jeden diagnostischen Schritt für sich hinterfragen muss: Ist er im Hinblick auf künftig zu erwartende steigende Fallzahlen skalierbar? Gewährleistet er noch die kurzen Bearbeitungszeiten, die heute von den Einsendern erwartet werden (vgl. Tab. 3)?
Diese grundsätzlichen Überlegungen sind umso wichtiger, als sich sehr viele gezielte Medikamente derzeit in der klinischen Prüfung befinden. Noch wissen wir nicht, welche erfolgreich sein und welche diagnostischen Tests sie erfordern werden. Doch eines ist sicher: Ohne den Nachweis der jeweiligen Zielstrukturen wird die Verwendung dieser neuen Medikamente weder sinnvoll noch finanziell vertretbar sein. Ihr Erfolg muss ebenso messbar überprüft werden, wie dies heute bei BCR-ABL1 selbstverständlich ist.
Aus dem Gesagten ergibt sich die durchaus praktische Schlussfolgerung, dass das handschriftliche Verfassen eines Befundes, so richtig dieser inhaltlich sein mag, nicht mehr dem Stand der Technik entspricht. Wir müssen uns tatsächlich sogar fragen lassen, inwieweit das Diktieren von Befunden ohne Softwareunterstützung – von der Spracherkennung bis zur künstlichen Intelligenz (KI) – auf Dauer Bestand haben kann.
Vorreiter ist hier die Radiologie, wo Algorithmen des maschinellen Lernens bereits routinemäßig zum Einsatz kommen, um die menschlichen Ressourcen richtig einzusetzen. Und außerhalb der Medizin erleben wir ohnehin, dass sich alles im Sinne des Internet of Things (IoT) vernetzt, um gewaltige Datenmengen zu sammeln und zu verarbeiten. Wenn also die maschinelle Mustererkennung und Entscheidungsunterstützung breiten Einzug in den Alltag hält, dann wäre es ein großes Versäumnis, diesen Technologiesprung nicht auch für die Leukämiediagnostik zu nutzen. Das, was jetzt nötig und möglich ist, hat in der Tat das Potenzial für eine disruptive Innovation.
Digitalisierung und KI
Wie muss man sich diese „Disruption“ vorstellen? Wird es tatsächlich zu einer völligen Umwälzung des diagnostischen Vorgehens kommen, bei der kein Stein auf dem anderen bleibt? Natürlich nicht. Wie bisher werden wir Informationen zum Phänotyp und Genotyp erfassen und dank ärztlicher Expertise zu einem Befund zusammenführen, am besten im Sinne eines „integrierten Befundes“, der die verschiedenen Einzelmethoden und ihre Ergebnisse differenziert berücksichtigt, zu einer Aussage verdichtet und kommentiert. Darauf werden auch weiterhin Diagnose, Klassifikation und häufig auch Prognose sowie Therapieentscheidung basieren.
Die Menge der Methoden und die Zahl der einzelnen Datenpunkte wird aber bald jedes ärztliche Vorstellungsvermögen übersteigen. Bereits heute erhalten wir bei der Immunphänotypisierung mit zehn parallelen Farben in unterschiedlichsten Kombinationen eine Datenflut, die ohne Softwareunterstützung nicht zu bewältigen wäre. Das Gleiche gilt für die Bilderfassung in der klassischen Chromosomenanalyse oder die Karyotypisierung. Speziell in der Molekulargenetik, also beim Nachweis von Veränderungen einzelner Gene bis hin zu ganzen Genomen, ist ohne Automatisierung und Digitalisierung keine sinnvolle Diagnostik mehr vorstellbar.
NGS und Bioinformatik
Seit das Next Generation Sequencing (NGS) Eingang in medizinische Labore gefunden hat, können wir prinzipiell sogar ganze Genome innerhalb von 40 Stunden sequenzieren. Dies ist zwar definitiv noch nicht Routine, aber dass das Whole Genome Sequencing bald routinetauglich sein wird, ist absehbar. Zum jetzigen Zeitpunkt fokussiert man sich noch eher auf einzelne Gene oder gezielte Genpanels, die man im Hochdurchsatz-Sequenzierverfahren deutlich schneller und preisgünstiger als früher analysieren kann.
NGS stellt eine Plattformtechnik dar, die es erlaubt, genomische Varianten unter Berücksichtigung der jeweiligen Fragestellung für jeden einzelnen Patienten zu analysieren. Daraus ergeben sich hohe Anforderungen speziell an die akkreditierten Prozesse im Labor (z. B. ISO 15189), aber mehr noch an die bioinformatische Datenverarbeitung und Auswertung. Im Gegensatz zu anderen Diagnoseverfahren für Leukämien und Lymphome ist die Bioinformatik beim NGS der Flaschenhals. Programme, die zu auswertbaren Daten führen, sind bereits etabliert, doch die anschließende Bewertung – auch im Hinblick auf Varianten mit unbekannter oder fehlender pathologischer Bedeutung – bedarf großer Sorgfalt und steht bei Weitem noch nicht vollautomatisiert und fehlerfrei zur Verfügung.
Umso mehr ist es jetzt unsere Aufgabe, genau diese große Menge an Information von einer reinen Analysemethode im wissenschaftlichen Setting zu einer Routinemethode mit patientenrelevanten Schlussfolgerungen zu bringen. Das erfordert neben massiver Softwareunterstützung auch eine komplexe Hardwarestruktur mit ausreichenden Sicherungen (auch im Sinne des Datenschutzes) sowie den Einsatz von künstlicher Intelligenz.
Die beiden Eckpunkte der „Leukämiediagnostik von morgen“ werden also auf der einen Seite durch die phänotypische Beschreibung des Blutausstriches und auf der anderen Seite durch den Einsatz von künstlicher Intelligenz beschrieben. Beide Extreme zusammenzubringen, wird im Zuge der immer detaillierteren Beschreibung einzelner Erkrankungen nötig sein. Im Umkehrschluss werden alle Methoden „vor NGS“ rückwirkend bezüglich ihrer Bedeutung, Aussagekraft und Sinnhaftigkeit auf dem Prüfstand stehen.
Mal abgesehen von der technisch-analytischen Reproduzierbarkeit der jeweiligen Messsysteme, die mehr oder minder erfahrungs- und personenabhängig zum hoffentlich richtigen Ergebnis kommen, ist vor allem die Frage zu stellen, wie wir bereits vorhandenes Wissen in die zukünftige Befunderstellung einfließen lassen können. Dies funktioniert nur mithilfe von großen Datenbanken und wissensbasierten Programmen, die die auswertenden MTAs und Ärzte, Molekularbiologen und Bioinformatiker unterstützen.
Chancen und Risiken
Das bedeutet somit, dass wir in der Leukämiediagnostik – paradigmatisch für viele weitere disruptive Schritte in der Medizin – gerade am Übergang vom Expertenwissen zu Befundungs-Algorithmen stehen. Dieser Wechsel wird nicht nur die Diagnostik in den nächsten fünf Jahren grundliegend verändern, sondern auch die Klassifikation und insbesondere die darauf aufbauenden Therapieentscheidungen für jeden einzelnen Patienten neu ausrichten.
Die darin liegenden Risiken, aber auch die darin enthaltenen Chancen nicht zu adressieren, wäre ein großer Fehler und weder für die einzelnen Ärzte und Patienten noch für die Weiterentwicklung des therapeutischen Fortschritts in der Hämato-Onkologie akzeptabel. Eine Integration der oben beschriebenen Möglichkeiten von NGS und KI in die heutige Goldstandard-Diagnostik so zu ermöglichen, dass vorhandenes Wissen konserviert und neues Wissen generiert werden kann – das ist jetzt unsere wichtigste Aufgabe. Die Leukämiediagnostik wird sich grundlegend verändern, und wir werden uns mit ihr verändern.
Autor
